All published articles of this journal are available on ScienceDirect.

Development, Validation and Application of RP-HPLC Method: Simultaneous Determination of Antihistamine and Preservatives with Paracetamol in Liquid Formulations and Human Serum
Authors Info & Affiliations
Abstract
In this article we describe development and validation of stability indicating, accurate, specific, precise and simple Ion-pairing RP-HPLC method for simultaneous determination of paracetamol and cetirizine HCl along with preservatives i.e. propylparaben, and methylparaben in pharmaceutical dosage forms of oral solution and in serum. Acetonitrile: Buffer: Sulfuric Acid (45:55:0.3 v/v/v) was the mobile phase at flow rate 1.0 mL min-1 using a Hibar® Lichrosorb® C18 column and monitored at wavelength of 230nm. The averages of absolute and relative recoveries were found to be 99.3%, 99.5%, 99.8% and 98.7% with correlation coefficient of 0.9977, 0.9998, 0.9984, and 0.9997 for cetirizine HCl, paracetamol, methylparaben and Propylparaben respectively. The limit of quantification and limit of detection were in range of 0.3 to 2.7 ng mL-1 and 0.1 to 0.8 ng mL-1 respectively. Under stress conditions of acidic, basic, oxidative, and thermal degradation, maximum degradation was observed in basic and oxidative stress where a significant impact was observed while all drugs were found almost stable in the other conditions. The developed method was validated in accordance with ICH and AOAC guidelines. The proposed method was successfully applied to quantify amount of paracetamol, cetirizine HCl and two most common microbial preservatives in bulk, dosage form and physiological fluid.
1. INTRODUCTION
Pharmaceutical liquid preparations are particularly vulnerable to microbial growth because of the nature of their ingredients. Such preparations are protected by the addition of preservatives that prevent alteration and degradation of product formulation [1]. Hence antibacterial and antifungal properties of preservatives i.e. parabens, make them an integral part of liquid dosage formulation. Parabens that include Methylparaben (MP Fig. (1)) and propylparaben (PP, Fig. (1)), are a group of the alkyl esters of p-hydroxybenzoic acid, have a low toxicity profile and a long history of use. MP and PP are the well-known preservatives that are used primarily for their bactericidal and fungicidal properties [2]. However usage of MP is toxic at higher concentrations and has an estrogenic effect [3]. The estrogenic activity of para-bens increases with the length of the alkyl group and it is believed that PP is estrogenic to a certain degree as well [4]; therefore finished product release specifications should include an identification test and content determination test with acceptance criteria and limits for each antimicrobial preservative present in the formulation [5, 6]. These indispensable requirements encourage our team to develop new stability indicating method for simultaneous estimation of APIs along with preservatives in liquid formulations to provide driving force in today’s pharmaceutical industry and clinical labs.
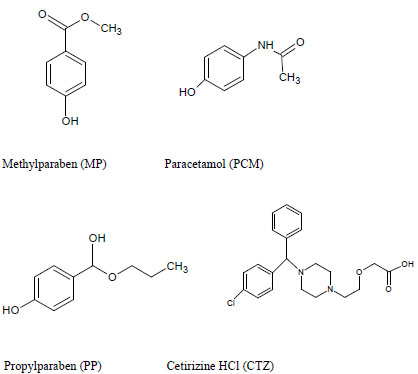
Cetirizine dihydrochloride (CTZ, Fig. 1) is a piperazine derivative and metabolite of hydroxyzine. It is described as a long acting non-sedating antihistamine and is used for the symptomatic relief of allergic conditions including rhinitis and chronic urticaria [7]. CTZ provides prompt relief to runny nose, itching of nose or throat due to respiratory allergies, urticaria. In some allergic circumstances it is essential to co-prescribe CTZ with paracetamol (PCM, Fig. 1), in conditions such as allergic rhinitis where antihistamines are the primary medication to be used [8]. PCM, a para-aminophenol derivative, has analgesic, antipyretic and weak anti-inflammatory activity [9]. PCM combination and co-administration with CTZ is often used as anti-allergic and antipyretic, and for the treatment of severe allergies and cold [10]. Majority of cold medicines contain several active ingredients that include antipyretics, analgesics, antitussive agents, mucolytic agents and antihistamines etc. [11]. such a combination of CTZ with PCM along with MP & PP is usually found in pediatric syrups. New analytical troubles are generated by the development of new pharmaceutical forms with classical active compounds. That is the case of oral solution containing PCM, CTZ with essential presence of MP and PP as preservatives.
Literature survey reveals some analytical methods are reported for simultaneous determination of paracetamol and cetirizine HCl by high-performance liquid chromatographic (HPLC) with isocratic elution [10, 12-14], with gradient HPLC method [15-17] and capillary zone electrophoresis [18], paracetamol and parabens by HPLC [19, 20], and the one and only method for CTZ with parabens (other than PP) [21], however our extensive research discloses that till now no stability indicating isocratic HPLC method is available which can determine the four ingredients concomitantly, therefore an endeavor was made to develop a simple, sensitive and validated stability indicating RP-HPLC method using UV detection to determine PCM and CTZ along with its essential microbial preservatives in liquid dosage forms and physiological fluid. Present work deals with method development, method validation and forced degradation study of the ingredients. The applicability of the method was confirmed for analysis in pharmaceutical products and body fluid studies. The results of analysis were validated in accordance with International Conference on Harmonisation (ICH) and Association of Official Analytical Chemists (AOAC) guidelines [22, 23].
2. MATERIALS AND METHOD
The proposed method is designed to be simple to use, sensitive, rapid with easy sample preparation for selected API’s and preservatives ingredients. Separation and quantification in pharmaceutical drug formulations and blood were achieved with an isocratic elution.
2.1. Reagents and Chemicals
Cetirizine HCl, paracetamol, methylparaben and propylparaben were kind gifts from national pharmaceuticals, whereas sulphuric acid, sodium heptane sulphonate, acetonitrile (HPLC grade) were purchased from Merck (Germany). From commercial source pharmaceutical dosage forms containing the respective ingredients were obtained including Novopyrin® (Specific Research Laboratories), Rigix Oral Solution® (AGP (Pvt) Ltd.), Linazine® (Alina Combine Pharmaceuticals (Pvt) Ltd.), and Pedrol®(Stanley Pharmaceuticals (Pvt) Ltd.).
2.2. Instruments
For chromatography a SIL 20AHT auto injector HPLC system comprising of CBM-20A controller, SPD 20A prominence UV/VIS detector, and Shimadzu LC 20 AT Prominence LC pump with software LC Solutions version 1.25 was used. Separation was performed on a Hibar® Lichrosorb® ODS C18 HPLC column, (4.6 x 250 mm; 10µm bead size) maintained at 25°C. A UV-visible Shimadzu 1650 PC spectrophotometer with UV Probe V2.0 software, ultrasonic cleaner (Elmasoni E 60 H), pH meter (Jenway 3240), shaker (Labnet Orbit 1000), centrifuge machine (HERMLE Z200A) and analytical balance (Sartorious TE2145) were used in the research work. Throughout the work only Class-A (Pyrex®) calibrated volumetric flasks and pipettes were used, as guided by Chan, Chung Chow et al. [24].
2.3. Chromatographic Conditions
HPLC analysis was carried out at ambient temperature. Compounds were chromatographed isocratically with a mobile phase consisting of acetonitrile: sodium n-heptane sulfonate 0.01M: sulfuric acid 0.1M (45:55:0.3 v/v/v) with apparent pH 3.0 ± 0.1 (pH adjusted if required using sulfuric acid 0.1M). Mobile phase was filtered by passing through a 0.45 µm membrane filter (Millipore, Bedford, MA, USA). Flow rate of mobile phase was 1.0 mL min-1, and injection volume of testing solution was 20µL. The effluent was monitored spectrophotometrically at wavelength of 230nm. Mobile phase was used as dilution solvent, throughout the analytical work.
3. EXPERIMENTAL
3.1. Standard Preparation
3.1.1. Stock Solutions
In a 100 mL class-A volumetric flask 15mg of PP reference standard was accurately weighed and dissolved in dilution solvent via sonication for few seconds (usually 10-15 seconds) to have a stock solution-A of 150 µg mL-1. Similarly in three other flasks 10mg, 135mg and 100mg of CTZ, MP and PCM were dissolved in diluent to get stock standard solutions of 100 µg mL-1, 1350 µg mL-1 and 1000 µg mL-1 concentration respectively. These stock solutions were further diluted for performing different experiments in the research work. For dilution and other solution preparation purposes calibrated class-A (Pyrex®) minimal volume pipette of 10 mL was used [24].
3.1.2. Lab. Prepared Mixture Solutions
For laboratory mixture preparation 15mg of PP reference standard was accurately weighed and dissolved in dilution solvent via sonication for few seconds to have a stock solution-A of 150 µg mL-1 . While in another class-A 100 mL volumetric flask, CTZ & MP reference standards were accurately weighed as 10mg and 13.5mg added by 10 mL of stock solution-A and dissolved in diluent and designated as stock solution-B. Similarly in another 100 mL volumetric flask, accurately 10mg of PCM was weighed and dissolve in 50 mL of dilution solvent; added by 10 mL of the stock solution-B and filled up to the mark with dilution solvent to get stock solution-C containing 10 µg mL-1 of CTZ, 100 µg mL-1 of PCM, and 13.5 µg mL-1 of MP and 1.5 µg mL-1 of PP. This working solution was used in further analytical steps.
3.2. Sample Preparation
3.2.1. Marketed Samples
For making a sample from syrups, one bottle of each brand was opened and content was mixed to get an evenly homogenized stock sample to contain 100 µg mL-1, 1350 µg mL-1, 1000 µg mL-1 and 150 µg mL-1 of CTZ, MP, and PCM & PP respectively. Sample volume of 10 mL was accurately taken in 100 mL volumetric flask and 50 mL of diluent was added. Then sample was mixed thoroughly by stirring for 10 minutes and added by diluent up to the mark and designated as stock sample-A; second dilution was prepared by dissolving 10 mL of stock sample-A in 100 mL diluent and then filtered through 0.45 µm filter and injected into HPLC system.
3.2.2. Sample for Serum
By a skilled clinical laboratory technician blood samples were collected from healthy volunteers in evacuated glass tube. Volunteers were confirmed to be not involved in any medication, smoking, and strenuous activity. In order to separate out serum, blood was shaken with the aid of shaker (Labnet Orbit 1000) at 200 rpm and centrifuged by centrifuge machine at 10,000 rpm for 15 minutes. Acetonitrile was added to serum (90:10, v/v) and centrifuged again at 10,000 rpm for 10 min to deprotonate it. The serum thus obtained was used for analysis and was stored at 20°C. For making working sample 10 mL of stock sample-A was taken in 100 mL flask, followed by 15 mL of serum. The sample thus obtained was stirred for 10 minutes and added with diluent up to the mark. Triplicate solutions were prepared for each working solution for analysis in serum, and were filtered through 0.45 µm filter prior to inject into HPLC system.
3.2.3. Forced Degradation Conditions
For this purpose 10 mL of stock solution-A was diluted in four individual 100 mL volumetric flasks and 15 mL of degrading agent were added to each flask individually, with exception of one to which only diluent was added; these included 0.1M HCL, 0.1M NaOH, 3%H2O2 and then to each flask diluent was added up to the mark. All the four samples were placed in water bath at 60°C for one hour. Samples were then filtered through 0.45 µm filter paper and injected into HPLC system.
3.2.4. Stability Studies
For stability studies commercially available samples were placed at accelerated conditions of temperature and humidity that is at 40°C with 75% relative humidity and at ambient conditions of 30°C temperature with 65% relative humidity in environmental chamber for six months. A stability protocol was followed for six months and assays were performed as described previously.
3.3. Method Validation
Method validation was performed following ICH and AOAC guidelines [22, 23] according to which assay validation was performed via various procedures including specificity, linearity, range, accuracy, intra-day & inter-day precision and system suitability etc. To study linearity, twenty dilutions were prepared from stock solution-B to give standard solutions in range of 10% to 200% of all ingredients content for marketed products. Standard calibration curve was generated using regression analysis. For specificity commonly used excipient in syrup preparation were spiked in a pre-weighed quantity of drugs and then peak areas were measured and calculated to determine the quantity of the drugs recovered. Precision was performed by analyzing corresponding standard daily for a period of three days (Inter-day precision or reproducibility), and three times a day with an interval of 08 hours (Intra-day precision or repeatability) against freshly prepared standard solutions. For determining accuracy reference standards were accurately weighed and added to previously analyzed syrup sample, to get three different concentration levels i.e. 110%, 120%, 130% of the API’s. At each level, samples were prepared in triplicate and recovery percentage was determined.
Limit of detection (LOD) and limit of quantification (LOQ) for the method were established by sequentially diluting standard solutions at decreasing concentrations, in range of 100 ng mL-1 to 0.01 ng mL-1 and injected onto chromatographic system. Limit of detection was defined as the concentration for which a signal-to-noise ratio of 3:1 was obtained and, for quantification limit; a signal-to-noise ratio of 10:1 was considered. Robustness was studied by analyzing same samples of syrup by deliberate variation in method parameters, such as in the chromatographic conditions, like mobile phase, pH, flow rate, temperature etc. System suitability of the method was evaluated by analyzing symmetry of the standard peaks, resolution and theoretical plates of the column.
4. RESULTS AND DISCUSSIONS
HPLC method development and its validation are the obligatory requirements for any drug available in the market to have high quality products. A few methods are available for determination of CTZ and PCM as described earlier, but many of them are used only for certain definite objectives and none can be generalized for simultaneous determination of CTZ and PCM with mentioned preservatives in pharmaceutical products and serum. In order to solve the problems of the gradient elution, we developed and validated an isocratic HPLC method for the simultaneous determination of the four ingredients.
Similarly none of the mentioned methods [10, 17] is as much sensitive as ours; in terms of its precision, accuracy, %recovery, limit of detection LOD and limit of quantification LOQ. Moreover this method is sensitive enough to be used for pharmacokinetic studies as its LOD & LOQ are in nano gram range, and provide best resolution of drugs. This section of the research work is part of our future article to be published soon afterwards.
4.1. Method Development & Optimization
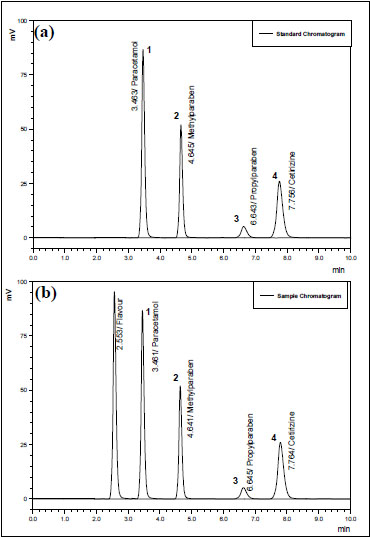
For developing a fit-to-purpose method for analysis, parameters, such as detection wavelength, mobile phase composition, optimum pH and concentration of the standard solutions were comprehensively studied. All the ingredients were diluted in dilution solvent and then run through UV spectrophotometer (Shimadzu 1650 PC) in UV range of 190nm-400nm to get maximal wavelengths, where maximum absorbance was attained i.e. 230nm, 220nm, and 254nm for CTZ, PCM and MP & PP respectively. However considering the difference of concentration of the ingredients and intensity of their absorbance a single wavelength method was adopted, that is 230nm at which all the molecules gave satisfactory absorbance. The chromatographic parameters were evaluated using a Hibar® Lichrosorb® C18 column. An ion-pairing mobile phase already developed by our research team [25] was selected in terms of its components & their proportions and was modified for better representable chromatography. The mobile phase composed of Acetonitrile: Buffer of previously mentioned proportion promoted a short run time (10 min) as shown in Fig. (2), so this condition was adopted in accompanying analysis.
4.2. Validation Studies
The linearity was determined in the range of 10% - 200% of claimed content for all API’s in marketed formulation. The assay was judged to be linear as the correlation coefficient was greater than 0.997 in all cases, calculated by the least-square method. A linear correlation was found between the peak areas and the concentrations, in the assayed range. The regression analysis data are presented in Table 1.
| Parameter | Inference | |||
|---|---|---|---|---|
| PCM | MP | PP | CTZ | |
| Linearity range (µg mL-1) | 10-200 | 1.35 27 | 0.15 - 3 | 1 - 20 |
| Correlation coefficient (r) | 0.9998 | 0.9984 | 0.9997 | 0.9977 |
| Regression equation (y=mx+c) Slope (m) | 0.096948 | 0.099248 | 0.096796 | 0.10153 |
| Intercept (c) | 0.140771 | -43487 | 0.153070 | -0.2371 |
| Limit of detection LOD (ng mL-1) | 0.5 | 0.8 | 0.1 | 0.5 |
| Limit of quantification (LOQ) (ng mL-1) | 2 | 2.7 | 0.3 | 2 |
Chromatogram shown in Fig. (2b) proves specificity or selectivity of the assayed method, as the chromatograms in samples were found identical with standard chromatogram (Fig. 2a) and no interference peak was observed either in pharmaceutical formulation or in physiological fluid. Peak purities higher than 97.0% (Tables 2 and 3) were obtained from assays of sample solutions, demonstrating that other compounds did not co-elute with peaks of interest. The chromatogram obtained with the mixture of the syrup excipient (Fig. 2b) proves that there is no interference from excipients thus peak of interest fulfills all the requirements of symmetrical peak, and hence the specificity is established.
Precision of an analytical method is the degree of coherence among individual test results when the method is applied repeatedly to multiple sampling of homogeneous bulk. Intra-day precision of the method was evaluated at three different independent concentrations i.e. 80%, 100%, and 120% for the drugs (n=3) by determining their assays. Inter-day precision of the method was tested for 3 days at the same concentration levels. Solutions for calibration curves were prepared every day on freshly basis. Since the inter-day and intra-day precision obtained %RSD was less than 2% it assures that the proposed method is quite precise and reproducible as shown in Table 2.
| Nominal concentration of active drugs (µg mL-1) | Day 1 | Day 2 | Day 3 | Mean | Inter-Day RSD% | |
|---|---|---|---|---|---|---|
| PCM | 40 | 99.8 | 99.2 | 99.5 | 99.51 | 0.33 |
| 50 | 100.9 | 98.2 | 99.3 | 99.47 | 1.36 | |
| 60 | 100.7 | 101.4 | 101.0 | 101.02 | 0.35 | |
| Mean | 100.48 | 99.59 | 99.93 | |||
| Inter-Day RSD% | 0.57 | 1.65 | 0.91 | |||
| MP | 1.08 | 99.5 | 98.9 | 99.3 | 99.23 | 0.31 |
| 1.35 | 100.6 | 99.1 | 99.7 | 99.80 | 0.76 | |
| 1.62 | 99.7 | 99.3 | 100.3 | 99.77 | 0.50 | |
| Mean | 99.93 | 99.10 | 99.77 | |||
| Inter-Day RSD% | 0.59 | 0.20 | 0.50 | |||
| PP | 0.12 | 98.7 | 99.1 | 97.9 | 98.57 | 0.62 |
| 0.15 | 99.6 | 98.5 | 98.1 | 98.73 | 0.79 | |
| 0.18 | 98.7 | 98.7 | 99.4 | 98.93 | 0.41 | |
| Mean | 99.00 | 98.77 | 98.47 | |||
| Inter-Day RSD% | 0.52 | 0.31 | 0.83 | |||
| CTZ | 0.8 | 98.7 | 99.1 | 99.5 | 99.10 | 0.40 |
| 1 | 99.3 | 99.7 | 98.8 | 99.27 | 0.45 | |
| 1.2 | 99.0 | 99.3 | 100.8 | 99.70 | 0.97 | |
| Mean | 99.00 | 99.37 | 99.70 | |||
| Inter-Day RSD% | 0.30 | 0.31 | 1.02 | |||
| Commercial Samples | Content (%) ± S.D.* | ||||
|---|---|---|---|---|---|
| PCM | MP | PP | CTZ | ||
| Rigix Oral Solution® | N/A | 98.2± 0.75 | 99.1± 0.39 | 98.7± 0.44 | |
| Linazine® | N/A | 97.17 ± 0.29 | 99.84 ± 0.76 | 98.79 ± 0.87 | |
| Pedrol[REMOVED HYPERLINK FIELD]® | 99.67± 0.95 | 100.59± 0.68 | 100.46± 0.74 | N/A | |
| Novopyrin ® | 100.88 ± 0.87 | 99.37 ± 0.85 | 98.65 ± 0.64 | N/A | |
Accuracy was investigated by means of addition of reference standards to a mixture of the syrup excipients. Recovery studies of the drug were carried out for the accuracy parameter at three different concentration levels i.e. multiple level recovery studies. A known amount of API’s standards were added into pre-analyzed sample and subjected to proposed HPLC method. The mean recovery (n = 9) was 99.0% - 99.7% (RSD=0.61%) for CTZ, 99.51% - 101.02% (RSD=0.68%) for PCM, 99.23% - 99.93% (RSD=0.52%) for MP and for PP 99.0% -98.47% (RSD=0.61%), demonstrating the accuracy of the method. Percentage recoveries for marketed products were found to be within the limits (Table 3).
The statistical analysis showed no significant difference between results obtained employing the analytical conditions established for the method and those obtained in the experiments in which variations of some parameters were introduced. The parameters used in system suitability test were symmetry of peaks, tailing factor, resolution and RSD of peak area for replicate syrup. Thus, the method showed to be robust for changes in mobile phase acetonitrile proportion, mobile phase pH, flow rate, and column temperature (Table 4).
| Chromatographic Conditions | Variation | Retention time (Minutes) | |||
|---|---|---|---|---|---|
| PCM | MP | PP | CTZ | ||
| Temprature (ºC) | 23 | 3.42 | 4.64 | 6.63 | 7.609 |
| 27 | 3.57 | 4.69 | 6.8 | 7.701 | |
| Flow rate (mL min-1) | 0.8 | 3.75 | 4.56 | 6.48 | 7.31 |
| 1.2 | 3.28 | 4.97 | 6.96 | 7.93 | |
| Vol. of Acetonitrile (%) | 43 | 3.67 | 4.71 | 6.75 | 7.83 |
| 47 | 3.37 | 4.54 | 6.56 | 7.55 | |
4.3. Forced Degradation Studies
Forced degradation is a process that involves degradation of drug products and drug substances at conditions more severe than accelerated conditions for stability and thus generates degradation products that can be studied to determine stability of the molecule. ICH guideline states that stress testing is intended to identify the likely degradation products which further helps in determination of the intrinsic stability of the molecule, establishing degradation pathways and to validate the stability indicating procedures used [22]. Forced degradation has to demonstrate specificity when developing stability-indicating methods and for this reason, it should be performed prior to implementation of stability studies to assure that analytical methods are stability-indicating [26], hence a forced degradation study was performed. During this study, upon treatment of CTZ, PCM, MP and PP with alkali (0.1M NaOH), acid (0.1M HCl), hydrogen peroxide (3%) and heat, it was observed that CTZ and PCM with preservatives are fairly stable to alkaline, oxidative and thermal stress conditions as the degradation was less than 10% that could be considered as allowed value for a product whose shelf life limits is in 90% - 110% range [27, 28]. In Fig. (3) samples showed different grades of degradation based on forced degradation conditions, in some cases higher than the individual components due to formation of some degradation products by combination of several of them.
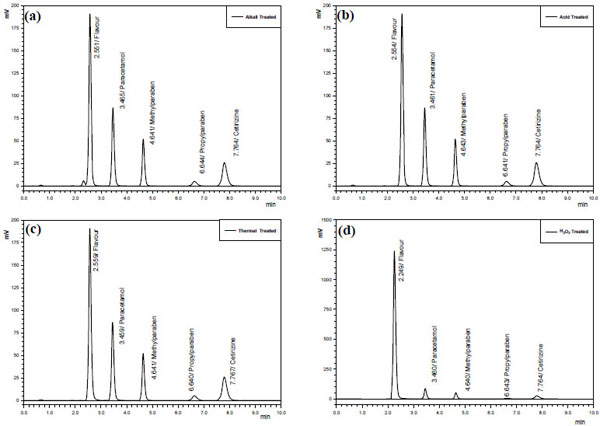
In our study PCM proved to have stability on thermal stress condition and no extra peak or increased area was observed which is in agreement with previous studies [29]. While oxidation with peroxide gave an extra peak, supposed to be benzoquinine-imine derivative (Fig. 3d), however, about 13% increased which is much more than observed by earlier researchers [26, 29] whereas about 4% decrease was observed for CTZ, MP and PP each under same stress condition. In alkaline conditions more PCM degradation was observed, which infers that greater oxidative degradation of PCM is attributed to oxidative vulnerability pushed up by electron withdrawing group in the form of amide as compared to hydrolysis. Whereas the other ingredients showed somewhat same behavior as in oxidative stress condition. A negligible degradation was observed for all the APIs (Fig. 3b) when acidic hydrolysis was performed which shows the same pattern as in the thermal stress condition. Esters of MP, PP and salt of carboxylic acid showed thermal stability which is similar to the results obtained in an earlier work [2]. Although parabens do possess carboxyl group but more drastic conditions are required to get them degraded as compared to PCM and CTZ. They have been found comparatively stable in all the provided conditions however as shown in the Fig. (3a and b) strong hydrolysis with both acid and base along with heating for longer time is required to decarboxylate the molecules to some extent, always not found on common experimental condition.
As shown by the chromatograms (Fig. 3) acidic and basic hydrolysis of paracetamol degraded peaks appear to be on the same retention time that can be assumed to be amino phenol which is the outcome of decarboxylation reaction after hydrolysis. The data in Table 5 also revealed that PCM and CTZ are usually more vulnerable to the stress conditions as compared to MP and PP. Further in Fig. (3a), base treated sample, degraded peaks of PCM is the Clear evidence of base hydrolysis of carboxylic group. Extremely high absorbance PCM in peroxide treated sample (Fig. 3d) showing the sensitivity of carboxylic salt being oxidized. The degraded peak appears to be on the same retaining time which can be assumed to be amino phenol and which is the outcome of decarboxylation reaction after hydrolysis.
| Stress conditions |
Time (min) |
Assay of active substance affected due to stress condition (%) | Assay of active substance under normal conditions (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| PCM | MP | PP | CTZ | PCM | MP | PP | CTZ | ||
|
Acid hydrolysis (0.1 M HCl) |
60 | 101.49 | 101.7 | 103.4 | 102.6 | 99.467 | 99.800 | 98.733 | 99.267 |
|
Base hydrolysis (0.1 M NaOH) |
60 | 110.276 | 102.243 | 104.335 | 105.096 | ||||
|
Oxidation (3% H2O2) |
60 | 113.784 | 96.722 | 96.245 | 96.688 | ||||
| Thermal (60ºC) | 60 | 100.53 | 105.47 | 104.38 | 105 | ||||
4.4. Stability Studies
Stability testing is an imperative part of the process of drug product development. Major purpose of stability testing is to provide documented data of how the quality of a drug substance or drug product varies with time under a variety of environmental conditions, for example temperature, humidity, and light and enables recommendation of storage conditions, retest periods, and shelf life to be established. It is a well-established fact that main aspects of drug product that play an important role in shelf-life determination are assay of the active drug and the degradation products generated during stability studies. Therefore it has become mandatory to perform stability studies of new drug moiety before filing in registration dossier. The stability studies include long term studies (12 months) and accelerated stability studies (6 months). But intermediate studies (6 months) can be performed at conditions milder than that used in accelerated studies [30]. The proposed assay method was applied to stability study of commercially available syrups, for which the samples were placed at specified condition and study was proceeded according to stability protocol as described in previous section (Table 6). Samples were analyzed and percentage of contents was measured. According to results obtained CTZ and PCM with preservatives were found to be stable at applied conditions of temperature and relative humidity, and were accurately analyzed with the proposed method.
| TEST (Claimed content) | INTERVAL | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Initial | 1Month | 2Month | 3Month | 4Month | 5Month | 6Month | Mean | RSD% | |
| Stability Studies at Accelerated (40ºC+75%H) | |||||||||
| PCM | 99.467 | 98.769 | 98.628 | 98.591 | 97.286 | 98.139 | 97.528 | 98.344 | 0.767 |
| MP | 99.800 | 99.576 | 99.783 | 100.546 | 99.826 | 98.763 | 98.375 | 99.524 | 0.732 |
| PP | 98.733 | 98.527 | 98.386 | 97.672 | 97.482 | 97.135 | 96.628 | 97.795 | 0.800 |
| CTZ | 99.267 | 99.626 | 99.295 | 99.386 | 98.492 | 99.184 | 98.783 | 99.148 | 0.388 |
| Stability Studies at Long Term (30ºC+65%H) | |||||||||
| PCM | 99.467 | 99.684 | 99.527 | 99.185 | 99.832 | 99.557 | 99.327 | 99.511 | 0.216 |
| MP | 99.800 | 99.536 | 99.638 | 99.318 | 99.107 | 98.836 | 98.937 | 99.310 | 0.369 |
| PP | 98.733 | 99.538 | 98.962 | 98.826 | 99.284 | 98.837 | 98.893 | 99.010 | 0.295 |
| CTZ | 99.267 | 99.538 | 99.294 | 99.628 | 99.192 | 98.963 | 98.882 | 99.252 | 0.276 |
CONCLUSION
A new HPLC method is proposed for simultaneous determination of cetirizine HCl, paracetamol with preservatives methylparaben and propylparaben in serum and liquid dosage forms. The proposed HPLC method described provides a simple, convenient and reproducible approach for simultaneous identification and quantification of four ingredients in human serum and pharmaceutical formulations with good separation and resolution. In addition, this method has the potential application to clinical research of drug combination. Analytical results are accurate and precise with good recovery and lowest detection limit values. In short, the developed method is simple, sensitive, easy, and efficient having short chromatographic time and can be used for routine analysis in QC laboratory and therapeutic monitoring. Due to the presence of different interferents in solution formulations such as coloring agents, flavoring agents, sweetening agents and preservatives other than PP and MP, the proposed method should be re-evaluated prior usage for commercial solution products containing ingredients other than those used in this work.
LIST OF ABBREVIATIONS
| (AOAC) | = Association of Official Analytical chemists |
| (CTZ) | = Cetirizine HCl |
| (HPLC) | = High Performance Liquid Chromatography |
| (ICH) | = International Conference on harmonization |
| (MP) | = Methylparaben |
| (PCM) | = Paracetamol |
| (PP) | = Propylparaben |
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
Declared none.
REFERENCES
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]

