All published articles of this journal are available on ScienceDirect.

Synthesis and Biological Evaluation of Novel Hybrid Molecules Containing Purine, Coumarin and Isoxazoline or Isoxazole Moieties
Authors Info & Affiliations
Abstract
Introduction:
The 1,3-dipolar cycloaddition reactions of nitrile oxides formed in situ (in the presence of NCS and Et3N) from the oximes of (purin-9-yl)acetaldehyde or (coumarinyloxy)acetaldehyde with allyloxycoumarins or 9-allylpurines, respectively resulted in 3,5-disubstituted isoxazolines. The similar reactions of propargyloxycoumarins or 9-propargylpurines led to 3,5-disubstituted isoxazoles by treatment with PIDA and catalytic amount of TFA.
Methods:
The new compounds were tested in vitro as antioxidant agents and inhibitors of soybean lipoxygenase LO, AChE and MAO-B.
Results:
The majority of the compounds showed significant hydroxyl radical scavenging activity. Compounds 4k and 4n presented LO inhibitory activity.
Conclusion:
Compound 13e presents an antioxidant significant profile combining anti-LO, anti-AChE and anti-MAO-B activities.
1. INTRODUCTION
Modified nucleosides [1, 2], coumarin derivatives [3-5], isoxazolines [6] and isoxazoles [7] represent classes of compounds with interesting broad range biological activities. Some modified nucleotides have been studied for the therapy of neurodegenerative disorders [8]. Coumarin derivatives have also been tested as acetylcholinesterase/ monoamine oxidase inhibitors for the treatment of Alzheimer’s Desease [9, 10].
In continuation to our recent studies on hybrid molecules with purine and coumarin moieties [11-13], on coumarin derivatives [3, 14, 15] and on modified nucleosides [16, 17], we present here the synthesis of new conjugated molecules as modified nucleosides, combining the coumarin and purine moieties through isoxazolines or isoxazoles as spacers. The new compounds were investigated for their antioxidant profile [free radical scavengers, lipid peroxidation and lipoxygenase (LO) inhibitors] as well as for their activity to ChEs and MAO enzymes searching for multipotent compounds.
2. MATERIALS AND METHODS
2.1. Chemistry
Some characteristic syntheses and selected data are given below:
2.1.1. General procedure. 1,3-Dipolar cycloaddition reactions of (purin-9-yl)acetaldehyde oximes with alkenyloxycoumarins. Synthesis of 4-methyl-6-({3-[(6-piperidin-1-yl-9H-purin-9-yl)methyl]-4,5-dihydroisoxazol-5-yl}methoxy)-2H-chromen-2-one (4a)
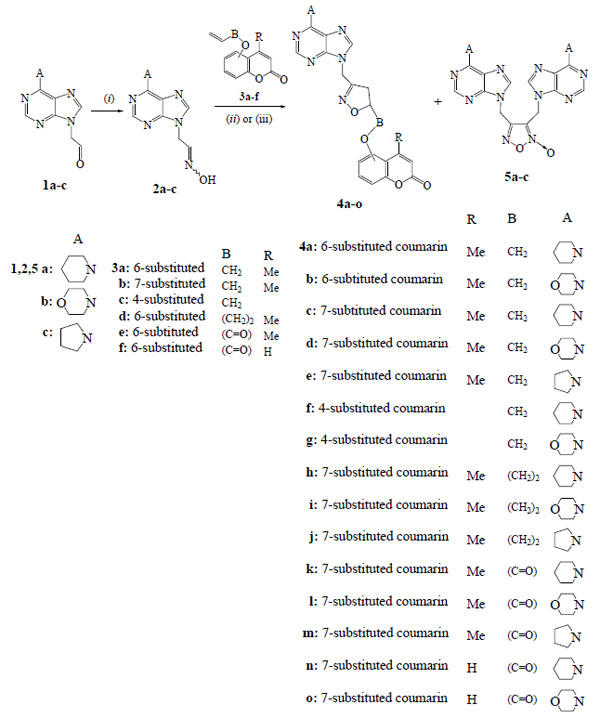
In the solution of oxime 2a (41 mg, 0.16 mmol) in dry DMF (5 ml) NCS (32 mg, 0.22 mmol) was added under stirring in portions during 1 h. The resulted mixture was stirred for 30 min. The allyloxycoumarin 3a (32 mg, 0.16 mmol) and Et3N (0.03 ml, 16 mg, 0.16 mmol) were then added and the mixture was stirred at r.t. for 24 h under N2 atmosphere. The mixture was filtered, the solid was washed with DCM and the filtrate was evaporated. The residue was chromatographed in a column [hexane/ethyl acetate (2:1)] and gave after the elution of starting coumarin 3a (5 mg, 16%) the isoxazoline 4a, 51 mg, (68% yield). White crystals, m.p.180-182°C (ethyl acetate); IR (Nujol): 3020, 1715, 1620, 1570 cm-1; 1H-NMR (CDCl3, 500 MHz) δ 1.63-1.80 (m, 6H), 2.40 (d, 3H, J=1.2 Hz), 2.98 (dd, 1H, J1=7.2 Hz, J2=17.1 Hz), 3.09 (dd, 1H, J1=11.2 Hz, J2=17.1 Hz), 4.05 (d, 2H, J=4.3 Hz), 4.17-4.29 (m, 4H), 4.93-5.05 (m, 1H), 5.15 (s, 2H), 6.29 (d, 1H, J=1.2 Hz), 6.99-7.05 (m, 2H), 7.23 (d, 1H, J=8.8 Hz), 7.80 (s, 1H), 8.32 (s, 1H); 13C-NMR (CDCl3, 125 MHz) δ 18.6, 24.7, 26.1, 37.5, 39.8, 45.6, 69.7, 78.6, 109.3, 115.7, 118.1, 119.2, 119.6, 120.6, 137.6, 150.6, 151.7, 152.6, 153.9, 154.3, 154.8, 160.7, 161.2; MS (ESI): m/z 475 [M+H]+, 497 ;[M+Na]+; Anal. Calcd (%) for C25H26N6O4: C, 63.28; H, 5.52; N, 17.71. Found: C, 63.17; H, 5.47; N, 17.86.
2.1.2. General Procedure. 1,3-Dipolar Cycloaddition Reactions of (Purin-9-yl)Acetaldehyde Oximes with Coumarinyl Acrylates. Synthesis of 4-Methyl-2-oxo-2H-chromen-6-yl 3-[(6-piperidin-1-yl-9H-purin-9-yl)methyl]-4,5-Dihydroisoxazole-5-Carboxylate (4k)
A solution of oxime 2a (62 mg, 0.24 mmol) in methanol (2 ml) was added dropwise during 1.5 h at r.t. in a mixture of acrylate 3e (60 mg, 0.26 mmol), PIDA (84 mg, 0.26 mmol) and TFA (4 μl, 5.7 mg, 0.05 mmol) in methanol (3 ml). The mixture was stirred for 4 h at r.t.. Then, the solvent was evaporated and the residue was separated by column chromatography [hexane/ethyl acetate (2:1)] to give the aldehyde 1a (10 mg, 17%) followed by the isoxazoline 4k, 83 mg (71% yield). White crystals, m.p.179-181°C (ethyl acetate); IR (KBr): 3027, 2912, 2834, 1719, 1702, 1621, 1589 cm-1; 1H-NMR (CDCl3, 500 MHz) δ 1.69-1.77 (m, 6H), 2.41 (s, 3H), 3.38-3.47 (m, 2H), 4.22-4.34 (m, 4H), 5.25 (s, 2H), 5.29 (dd, 1H, J1=6.9 Hz, J2=10.7 Hz), 6.32 (s, 1H), 7.21-7.28 (m, 2H), 7.36 (d, 1H, J=8.7 Hz), 7.81 (s, 1H), 8.35 (s, 1H); 13C-NMR (CDCl3, 125 MHz) δ 18.7, 24.5, 26.1, 39.4, 39.8, 46.6, 78.2, 116.0, 117.0, 118.2, 119.3, 120.7, 124.7, 137.8, 146.0, 149.1, 150.7, 151.3, 151.5, 153.3, 154.1, 160.1, 167.9; MS (ESI): m/z 489 [M+H]+; Anal. Calcd (%) for C25H24N6O5: C, 61.47; H, 4.95; N, 17.20. Found: C, 61.58; H, 4.92; N, 17.12.
2.1.3. General procedure. 1,3-Dipolar cycloaddition reactions of (coumarinyl)acetaldehyde oximes with 9-allylpurines. Synthesis of 4-methyl-6-({5-[(6-piperidin-1-yl-9H-purin-9-yl)methyl]-4,5-dihydroisoxazol-3-yl}methoxy)-2H-chromen-2-one (11a)
In the solution of oxime 9a (37 mg, 0.16 mmol) in dry DMF (5 ml) NCS (32 mg, 0.22 mmol) was added under stirring in portions during 1 h. The resulted mixture was stirred for 30 min. The allylpurine 10a (39 mg, 0.16 mmol) and Et3N (0.03 ml, 16 mg, 0.16 mmol) were then added and the mixture was stirred at r.t. for 24 h under N2 atmosphere. The mixture was filtered, the solid was washed with DCM and the filtrate was evaporated. The residue was chromatographed in a column [hexane/ethyl acetate (2:1)] and gave after the elution of starting purine 10a (5 mg, 13%) the isoxazoline 11a, 53 mg, (70% yield). White crystals, m.p.148-150°C (ethyl acetate); IR (Nujol): 3080, 1720, 1630, 1565 cm-1; 1H-NMR (CDCl3, 500 MHz) δ 1.69-1.79 (m, 6H), 2.40 (s, 3H), 3.07 (dd, 1H, J1=6.6 Hz, J2=17.9 Hz), 3.24 (dd, 1H, J1=10.9 Hz, J2=17.9 Hz), 4.23-4.33 (m, 4H), 4.39 (dd, 1H, J1=5.2 Hz, J2=14.0 Hz), 4.46 (dd, 1H, J1=5.4 Hz, J2=14.0 Hz), 4.76 (s, 2H), 5.03-5.14 (m, 1H), 6.31 (s, 1H), 6.97-7.03 (m, 2H), 7.25 (d, 1H, J=8.5 Hz), 7.88 (s, 1H), 8.37 (s, 1H); 13C-NMR (CDCl3, 125 MHz) δ 18.7, 24.0, 26.3, 38.3, 45.6, 49.3, 63.2, 78.6, 109.4, 114.9, 116.0, 118.3, 119.0, 120.8, 124.4, 138.9, 145.6, 149.6, 151.8, 152.3, 154.0, 160.3, 162.1; MS (ESI): m/z 475 [M+H]+, 497 [M+Na]+; Anal. Calcd (%) for C25H26N6O4: C, 63.28; H, 5.52; N, 17.71. Found: C, 63.35; H, 5.47; N, 17.59.
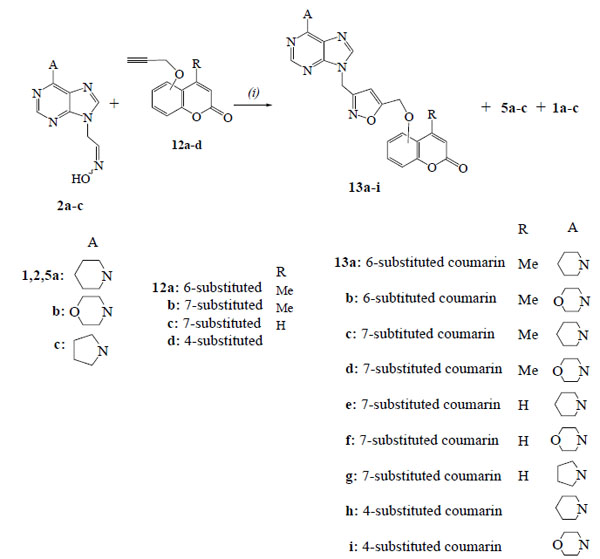
2.1.4. General procedure. 1,3-Dipolar cycloaddition reactions of (purin-9-yl)acetaldehyde oximes with propargyloxycoumarins. Synthesis of 4-methyl-6-({3-[(6-piperidin-1-yl-9H-purin-9-yl)methyl]isoxazol-5-yl}methoxy)-2H-chromen-2-one (13a)
TFA (4 μl, 5.7 mg, 0.05 mmol) was added to the solution of propargyloxycoumarin 12a (56 mg, 0.26 mmol) and PIDA (84 mg, 0.26 mmol) in methanol (3 ml). Then, in the resulted mixture, a solution of oxime 2a (62 mg, 0.24 mmol) in methanol (2 ml) was transferred dropwise during 1.5 h and the mixture was stirred at r.t. for 4 h. The solvent was evaporated and the solid residue was separated by column chromatography [hexane/ethyl acetate (2:1)] followed by PTLC (ethyl acetate) to give the aldehyde 1a (5 mg, 9%) and the isoxazole 13a (73 mg, 64%). White crystals, m.p.151-152°C (DCM); IR (Nujol): 3030, 1710, 1620, 1570 cm-1; 1H-NMR (CDCl3, 500 MHz) δ 1.68-1.82 (m, 6H), 2.40 (s, 3H), 4.21-4.38 (m, 4H), 5.16 (s, 2H), 5.48 (s, 2H), 6.32 (s, 1H), 6.44 (s, 1H), 7.06-7.17 (m, 2H), 7.29 (d, 1H, J=8.7 Hz), 7.83 (s, 1H), 8.40 (s, 1H); 13C-NMR (CDCl3, 125 MHz) δ 18.6, 26.2, 29.7, 38.9, 47.2, 62.1, 103.4, 109.8, 116.0, 118.3, 119.3, 119.6, 120.8, 138.1, 148.9, 150.1, 151.5, 152.2, 154.0, 159.6, 160.5, 160.7, 168.8; MS (ESI): m/z 473 [M+H]+, 495 [M+Na]+; Anal. Calcd (%) for C25H24N6O4: C, 63.55; H, 5.12; N, 17.79. Found: C, 63.62; H, 5.17; N, 17.63.
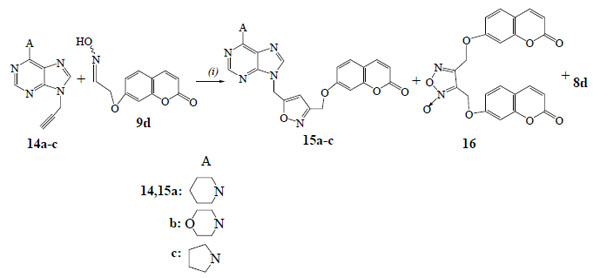
2.1.5. General procedure. 1,3-Dipolar cycloaddition reactions of [(2-oxo-2H-chromen-7-yl)oxy]acetaldehyde oxime (9d) with propargylpurines. Synthesis of 7-({5-[(6-piperidin-1-yl-9H-purin-9-yl)methyl]isoxazol-3-yl}methoxy)-2H-chromen-2-one (15a)
TFA (4 μl, 5.7 mg, 0.05 mmol) was added to the solution of propargylpurine 14a (63 mg, 0.26 mmol) and PIDA (84 mg, 0.26 mmol) in methanol (3 ml). Then, in the resulted mixture, a solution of oxime 9d (53 mg, 0.24 mmol) in methanol (2 ml) was transferred dropwise during 1.5 h and the mixture was stirred at r.t. for 4 h. The solvent was evaporated and the solid residue was separated by column chromatography [hexane/ethyl acetate (2:1)] followed by PTLC (ethyl acetate) to give the aldehyde 8d (5 mg, 11%) and the isoxazole 15a (62 mg, 56%). White crystals, m.p. 140-142°C (DCM); IR (Nujol): 3040, 1725, 1595 cm-1; 1H-NMR (CDCl3, 500 MHz) δ 1.65-1.77 (m, 6H), 4.18-4.35 (m, 4H), 5.16 (s, 2H), 5.52 (s, 2H), 6.25 (d, 1H, J=9.5 Hz), 6.41 (s, 1H), 6.80-6.93 (m, 2H), 7.36 (d, 1H, J=9.2 Hz), 7.60 (d, 1H, J=9.5 Hz), 7.85 (s, 1H), 8.37 (s, 1H); 13C-NMR (CDCl3, 125 MHz) δ 24.7, 26.2, 38.7, 47.0, 62.0, 102.3, 103.1, 112.7, 113.5, 114.0, 119.6, 129.1, 137.8, 143.1, 149.7, 151.7, 153.4, 155.9, 160.4, 160.7, 161.0, 166.8; MS (ESI): m/z 459 [M+H]+, 497 [M+K]+; Anal. Calcd (%) for C24H22N6O4: C, 62.87; H, 4.84; N, 18.33. Found: C, 62.93; H, 4.78; N, 18.17.
2.1.6. General procedure. 1,3-Dipolar cycloaddition reactions of [(2-oxo-2H-chromen-7-yl)oxy]acetaldehyde oxime (9d) with vinylpurines. Synthesis of 7-{[5-(6-piperidin-1-yl-9H-purin-9-yl)-4,5-dihydroisoxazol-3-yl]methoxy}-2H-chromen-2-one (18a)
TFA (4 μl, 5.7 mg, 0.05 mmol) was added to the solution of vinylpurine 17a (60 mg, 0.26 mmol) and PIDA (84 mg, 0.26 mmol) in methanol (3 ml). Then, in the resulted mixture, a solution of oxime 9d (53 mg, 0.24 mmol) in methanol (2 ml) was transferred dropwise during 1.5 h and the mixture was stirred at r.t. for 4 h. The solvent was evaporated and the solid residue was separated by column chromatography [hexane/ethyl acetate (2:1)] followed by PTLC (ethyl acetate) to give the aldehyde 8d (7 mg, 15%) and the isoxazoline 18a (71 mg, 66%). White crystals, m.p. 197-199°C (ethyl acetate); IR (Nujol): 3040, 1710, 1585 cm-1; 1H-NMR (CDCl3, 500 MHz) δ 1.67-1.81 (m, 6H), 3.68-3.75 (m, 2H), 4.22-4.34 (m, 4H), 5.06 (d, 1H, J=12.8 Hz), 5.12 (d, 1H, J=12.8 Hz), 6.29 (d, 1H, J=9.6 Hz), 6.78-6.85 (m, 1H), 6.89-6.95 (m, 2H), 7.41 (d, 1H, J=9.1 Hz), 7.63 (d, 1H, J=9.6 Hz), 7.77 (s, 1H), 8.29 (s, 1H); 13C-NMR (CDCl3, 125 MHz) δ 24.7, 26.2, 41.7, 46.9, 63.1, 84.4, 102.5, 112.4, 113.7, 114.3, 120.3, 129.2, 136.0, 143.0, 149.7, 152.0, 153.4, 155.8, 155.9, 160.6, 160.7; MS (ESI): m/z 447 [M+H]+; Anal. Calcd (%) for C23H22N6O4: C, 61.87; H, 4.97; N, 18.82. Found: C, 61.95; H, 4.93; N, 18.72.
2.2. Biology
2.2.1. Materials and Methods
All the reagents used were commercially available by Merck, 1,1-diphenyl-2-picrylhydrazyl (DPPH), nordihydroguairetic acid (NDGA) were purchased from the Aldrich Chemical Co. Milwaukee, WI, (USA). Soybean Lipoxygenase, linoleic acid sodium salt, were obtained from Sigma Chemical, Co. (St. Louis, MO, USA). Trolox were purchased by Fluka A.G. For in vitro determination a UV-Vis Shimadzu Spectrophotometer was used.
2.2.2. In vitro
In the in vitro assays each experiment was performed at least in triplicate and the standard deviation of absorbance was less than 10% of the mean.
2.2.2.1. Determination of the reducing activity of the stable radical 1, 1-diphenyl-picrylhydrazyl (DPPH) [14]
To a solution of DPPH (100μM) in absolute ethanol an equal volume of the compounds dissolved in ethanol was added. As control solution ethanol was used. The concentration of the solutions of the compounds was 100μM. After 20 and 60 min at room temperature the absorbance was recorded at 517 nm (Table 5). NDGA was used as a standard.
2.2.2.2. Competition of the tested compounds with DMSO for hydroxyl radicals [37]
The hydroxyl radicals generated by the Fe 3+ /ascorbic acid system, were detected according to Nash, by the determination of formaldehyde produced from the oxidation of DMSO. The reaction mixture contained EDTA (0.1 mM), Fe 3+ (167 μM), DMSO (33 mM) in phosphate buffer (50 mM, pH 7.4), the tested compounds (concentration 0.1mM) and ascorbic acid (10 mM). After 30 min of incubation (37°C) the reaction was stopped with CCl3COOH (17% w/v) (Table 5). Trolox was used as a standard.
2.2.2.3. Inhibition of linoleic acid lipid peroxidation [14]
Production of conjugated diene hydroperoxide by oxidation of linoleic acid sodium salt in an aqueous solution was monitored at 234 nm. 2,2’-Azobis(2-amidinopropane) dihydrochloride (AAPH) was used as a free radical initiator. 10 μl of the 16 mM linoleic acid sodium salt solution was added to the UV cuvette containing 0.93 ml of 0.05 M phosphate buffer, pH 7.4 prethermostated at 37°C. The oxidation reaction was initiated at 37°C under air by the addition of 50 μl of 40 mM AAPH solution. Oxidation was carried out in the presence of 10 μl of the examined compounds (stock solution in DMSO). In the assay without antioxidant, lipid oxidation was measured in the presence of the same level of DMSO. The rate of oxidation at 37°C was monitored by recording the increase in absorption at 234 nm caused by conjugated diene hydroperoxides (Table 5). Trolox was used as a standard.
2.2.2.4. Soybean lipoxygenase inhibition study in vitro [14]
In vitro study was evaluated as reported previously. The tested compounds dissolved in ethanol were incubated at room temperature with sodium linoleate (0.1 mM) and 0.2 ml of enzyme solution (1/9 x10-4 w/v in saline). The conversion of sodium linoleate to 13-hydroperoxylinoleic acid at 234 nm was recorded and compared with the appropriate standard inhibitor nordihydroguaretic acid (IC50 5.5 μM). Several concentrations were used for the determination of IC50 values (Table 5).
2.2.2.5. Inhibition Study on ChEs in vitro
In vitro inhibition of electric eel acetylcholinesterase (eeAChE; 463 U/mg, Sigma) and equine serum butyrylcholinesterase (esBChE; 13 U/mg, Sigma) was investigated with a 96-well plate procedure based on the classical Ellman’s spectrophotometric test, as already described [35].
2.2.2.6. Inhibition Study on MAOs in vitro
Inhibition of rat monoamine oxidase A and B was studied by means of a spectrofluorimetric method as previously detailed [38].
3. RESULTS AND DISCUSSION
3.1. Chemistry
The reactions studied and the title new compounds received are depicted in Schemes (1-5). The nitrile oxide, generated from the oxime 2a [18] by chlorination with NCS in DMF solution followed by addition of Et3N in an one-pot procedure, was treated with the allyloxycoumarin 3a [19] under stirring for 24 h and Ar atmosphere (Scheme 1) to give the isoxazoline 4a in 68% yield (Table 1, entry 1). The isoxazoline 4a has the expected regiochemistry [18] as indicated by HMBC experiments. There is correlation between the protons of NCH2 group [5.15 ppm (s, 2H)] with the carbon of 4-CH2 (isoxazoline) (37.5 ppm in 13C-NMR) and not with the 5-CH (isoxazoline) (78.6 ppm).
| Entry | Oxime | Alkenyloxycoumarin | Product (Yield %) |
|---|---|---|---|
| 1 | 2a | 3a | 4a (68) |
| 2 | 2b | 3a | 4b (68) |
| 3 | 2a | 3b | 4c (66) |
| 4 | 2b | 3b | 4d (71) |
| 5 | 2c | 3b | 4e (69) |
| 6 | 2a | 3c | 4f (73) |
| 7 | 2b | 3c | 4g (70) |
| 8 | 2a | 3d | 4h (64) |
| 9 | 2b | 3d | 4i (65) |
| 10 | 2c | 3d | 4j (67) |
| 11 | 2a | 3e | 4k (71)*, 1a (17%) |
| 12 | 2b | 3e | 4l (68)*, 1b (14%) |
| 13 | 2c | 3e | 4m (65)*, 1c (14%) |
| 14 | 2a | 3f | 4n (65)*, 1a (15%) |
| 15 | 2b | 3f | 4o (67)*, 1b (12%) |
The analogous reaction of the 6-allyloxycoumarin 3a with the nitrile oxide formed in situ from the oxime 2b (prepared by treatment of aldehyde 1b [13] in ethanol-water with NH2OH.HCl in the presence of anhydrous CH3COONa at 80°C for 2.5 h) resulted in the isoxazoline 4b in 68% yield (Table 1, entry 2). The 1,3-dipolar cycloaddition reactions of 7-allyloxycoumarin 3b [37, 38] with nitrile oxides resulted from the oximes 2a,b and 2c (synthesized from the aldehyde 1c [13]) gave the isoxazolines 4c,d,e respectively (Table 1, entries 3-5). The one-pot reactions of 4-allyloxycoumarin (3c) [12, 20] with the nitrile oxides of oximes 2a,b led to the isoxazolines 4f,g in 73% and 70% yield respectively (Table 1, entries 6,7). The isoxazolines 4h-j isolated from the reactions of 7-butenyloxycoumarin 3d [12] with the nitrile oxides formed from oximes 2a-c respectively (Table 1, entries 8-10). The isoxazoline 4h has the same regiochemistry, like the others, as the protons of NCH2 group [5.16 ppm (s, 2H)] in HMBC experiments are correlated with the carbon of 4-CH2 (isoxazoline) (38.7 ppm in 13C-NMR) and not with the 5-CH (isoxazoline) (78.9 ppm). In all the above experiments none of the possible furoxans 5a-c was detected.
In the case of (coumarin-6-yl)acrylate 3e [12] the one-pot procedure with NCS, Et3N and oxime 2a led to the isoxazoline 4k in only 34% yield along with the furoxan 5a (27%) [18]. When the reaction of acrylate 3e with the oxime 2a was performed with PIDA as oxidizing agent in the presence of catalytic amount of TFA in methanol under stirring for 4 h, the yield for the isoxazoline 4k increased to 71% (Table 1 entry 11). No furoxan 5a was detected. The reactions of acrylates 3e,f with the oximes 2b,c and 2a,b respectively in the presence of PIDA and TFA gave the isoxazolines 4l-o (Table 1, entries 12-15). No furoxans 5b,c were detected by this method.
Another way is presented in Scheme (2) for the synthesis of the hybrid compounds 11a-g using the 1,3-dipolar cycloaddition reactions of nitrile oxides, formed in situ from the (coumarinyloxy)acetaldehyde oximes 9a-d, with the 9-allylpurines 10a-d. The oximes 9a-d (84-88% yields) were prepared for first time from the corresponding substituted acetaldehydes 8a-d [21, 22] by treatment with NH2OH.HCl in ethanol-water in the presence of anhydrous CH3COONa at 80° C for 1.5 h. The acetaldehydes 8a-d were in turn prepared in 87-94% yields by refluxing a hydrochloric acid solution of the corresponding acetals 7a-d for 1 h. The acetals 7a-d were synthesized from the hydroxycoumarins 6a-d after heating with 2-bromo-1,1-diethoxyethane and K2CO3 in DMF at 90°C for 24 h.
The reaction of 9-allylpurine 10a with the nitrile oxide, resulted from the oxime 9a, was carried out by the above described one-pot procedure with NCS and Et3N and led to the isoxazoline 11a in 70% yield (Table 2, entry 1). In the case of isoxazoline 11a the coumarin moiety is connected in the 3-position of the isoxazoline ring and the purine moiety in the 5-position, differentiated from the isoxazoline 4a. The regiochemistry of 11a is demonstrated by HMBC experiments as there is correlation of the protons of OCH2 group with the carbon of 4-CH2 (isoxazoline) (38.3 ppm in 13C-NMR) and not with the 5-CH (isoxazoline) (78.6 ppm).
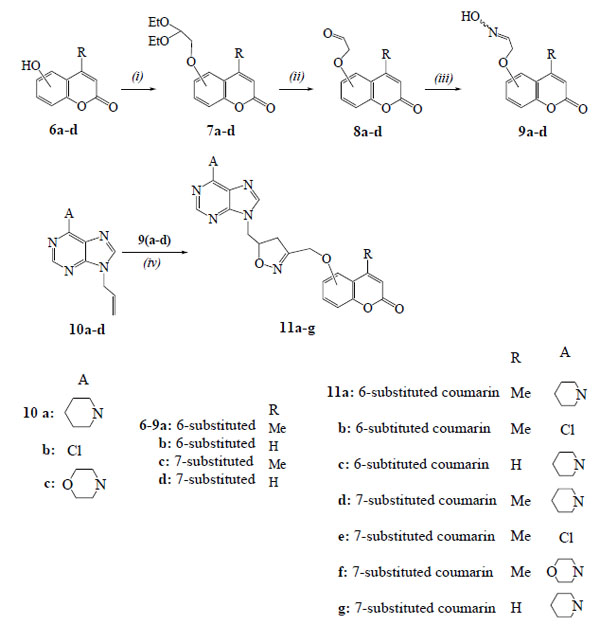
The isoxazolines 11b,c were isolated in 67% and 70% yields respectively from the reactions of 9-allylpurines 10b,a with the nitrile oxides synthesized from the oximes 9a,b (Table 2, entries 2,3). The analogous reactions of the oxime 9c with the purines 10a-c in the presence of NCS and Et3N gave the isoxazolines 11d-f (Table 2, entries 4-6) (Scheme 2). The isoxazoline 11g was obtained from the reaction of nitrile oxide, produced from the oxime 9d, with the purine 10a (Table 2, entry 7).
| Entry | (Coumarinyloxy)acetaldehyde oxime | 9-Allylpurine | Product (Yield %) |
|---|---|---|---|
| 1 | 9a | 10a | 11a (70) |
| 2 | 9a | 10b | 11b (67) |
| 3 | 9b | 10a | 11c (70) |
| 4 | 9c | 10a | 11d (72) |
| 5 | 9c | 10b | 11e (68) |
| 6 | 9c | 10c | 11f (65) |
| 7 | 9d | 10a | 11g (64) |
We examined next the reactions of nitrile oxides generated from the oximes 2a-c with the propargyloxycoumarins 12a-d in order to obtain the hybrids 13a-i with isoxazole ring (Scheme 3). The reaction of propargyloxycoumarin 12a [23] with the nitrile oxide, resulted from the oxime 2a under the above described one-pot procedure (NCS, Et3N), gave the expected product 13a only in 28% yield (Table 3, entry 1). In order to increase the yield of this 1,3-dipolar cycloaddition reaction, we investigated the best reaction conditions using different oxidants and solvents under different temperatures (Table 3).
| Entry | Reactants (mmol) | Solvent | T (°C) | Products (Yield %) |
|---|---|---|---|---|
| 1 | NCS (1.4), Et3N (1) | DMF | 25 | 13a (28), 5a (25), 2a (45) |
| 2 | PIDA (1.1) | MeOH | 25 | 13a (37), 5a (21), 1a (12) |
| 3 | PIDA (1.1), TFA (0.2) | MeOH | 25 | 13a (64), 5a (16), 1a (9) |
| 4 | PIDA (1.1), TFA (0.2) | MeOH | 0 | 13a (32), 5a (45) |
| 5 | PIDA (1.1), TFA (0.2) | MeOH | 60 | 13a (19), 5a (23), 1a (42) |
| 6 | PIDA (1.1), TFA (0.2) | MeOH/H2O | 25 | 13a (56), 5a (19), 1a (15) |
| 7 | PIDA (1.1), TFA (0.2) | DCM | 25 | 13a (34), 5a (48), 1a (12) |
| 8 | PIDA (1.1), TFA (0.2) | THF | 25 | 13a (25), 5a (52), 1a (19) |
| 9 | PIDA (1.1), TFA (0.6) | MeOH | 25 | 13a (18), 5a (27), 1a (48) |
| 10 | PIDA (2), TFA (0.6) | MeOH | 25 | 13a (14), 5a (31), 1a (37) |
| 11 | PIFA (1) | MeOH | 25 | 13a (45), 5a (39), 1a (15) |
PIDA as oxidant of oxime 2a, for the formation of the corresponding nitrile oxide in MeOH under r.t. gave a little better yield for the product 13a (37%) (Table 3, entry 2). The method with PIDA and a catalytic [18] amount of TFA was the best with the yield for 13a to increase (64%). By changing the temperature to 0°C or 60°C the yields for 13a were decreased, while the amount of furoxan 5a was increased and the hydrolysis product, aldehyde 1a, was the major product at 60°C (Table 3, entries 4,5). When MeOH/water was used as solvent, the yield of 13a was a little lower (Table 3, entry 6), while the DCM or the THF led to furoxan 5a as the major product (Table 3, entries 7 or 8 respectively). The increase in the amount of TFA or the PIDA gave a larger amount of the aldehyde 1a (Table 3, entries 9 or 10 respectively). The PIFA, which was the oxidant of choice for analogous reactions without solvent [18], led to lower yield of isoxazole 13a (Table 3, entry 11).
After the examination of suitable reaction conditions, the propargyloxycoumarins were reacted with the nitrile oxides generated from the oximes 2a-c in the presence of PIDA and catalytic amount of TFA (Scheme 3, Table 4). The reactions of 4-methyl-6-propargyloxycoumarin (12a) with the oximes 2a,b led to the isoxazoles 13a,b respectively (Table 4, entries 1,2). The isoxazole 13a has the expected regiochemistry [18] as indicated by HMBC experiments. There is a correlation between the protons of NCH2 group [5.48 ppm (s, 2H)] with the carbon of 4-CH (isoxazole) (103.4 ppm in 13C-NMR) and not with the 5-C (isoxazole) (168.8 ppm). The isoxazoles 13c,d were received from the reactions of 4-methyl-7-propargyloxycoumarin (12b) [24] with the oximes 2a,b (Table 4, entries 3,4). The analogous reactions of 7-propargyloxycoumarin (12c) [25] with the oximes 2a-c led to the isoxazoles 13e-g (Table 4, entries 5-7). The isoxazoles 13h,i were isolated from the reactions of 4-propargyloxycoumarin (12d) [26] with the oximes 2a,b (Table 4, entries 8,9).
| Entry | Oxime | Propargyloxycoumarin | Product (Yield %) |
|---|---|---|---|
| 1 | 2a | 12a | 13a (64), 5a (16), 1a (9) |
| 2 | 2b | 12a | 13b (62), 5b (18), 1b (9) |
| 3 | 2a | 12b | 13c (57), 5a (19), 1a (10) |
| 4 | 2b | 12b | 13d (54), 5b (19), 1b (12) |
| 5 | 2a | 12c | 13e (56), 5a (19), 1a (10) |
| 6 | 2b | 12c | 13f (53), 5b (19), 1b (11) |
| 7 | 2c | 12c | 13g (53), 5c (18), 1c (11) |
| 8 | 2a | 12d | 13h (60), 5a (16), 1a (10) |
| 9 | 2b | 12d | 13i (58), 5b (16), 1b (10) |
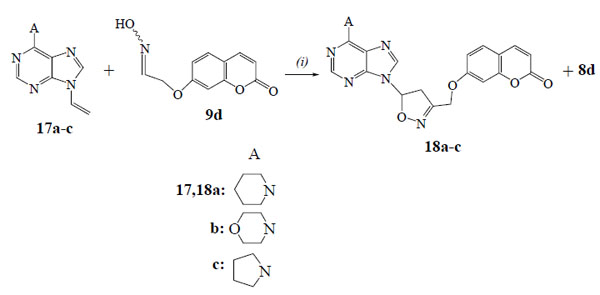
We studied also the reactions of oxime 9d with the 9-propargylpurines 14a-c (Scheme 4) and the 9-vinylpurines 17a-c (Scheme 5) in the presence of PIDA and catalytic amount of TFA. From the reaction of purine 14a the isoxazole 15a (56%) was isolated along with the dimerization product, furoxan 16 (19%). The regiochemistry of 15a is demonstrated by HMBC experiments as there is correlation of the protons of OCH2 group with the carbon of 4-CH (isoxazole) (103.1 ppm in 13C-NMR) and not with the 5-C (isoxazole) (166.8 ppm). The reactions of purines 14b,c led to the isoxazoles 15b (59%) and 15c (53%) respectively, while the furoxan 16 (19% and 17%) was also formed. The above resulted isoxazoles 15a-c were formed despite the possibility for isomerization of alkynes 14a-c to the corresponding allenes [27].
The 9-vinylpurine 17a [28] reacted with the oxime 9d to give the isoxazoline 18a (66%) (Scheme 5). No furoxan 16 was detected in the reaction mixture. The isoxazoline 18a has the same regiochemistry, like the others, as the protons of OCH2 group [5.06 ppm (d, 1H)/5.12 ppm (d, 1H)] in HMBC experiments are correlated with the carbon of 4-CH2 (isoxazoline) (46.9 ppm in 13C-NMR) and not with the 5-CH (isoxazoline) (84.4 ppm). The reactions of purines 17b,c gave the isoxazolines 18b (62%) and 18c (60%) respectively, along with the aldehyde 8d (15%).
3.2. Biological Evaluation
The formation of Reactive Oxygen Species (ROS) is a consequence of cell metabolism for aerobic organisms. Due to the extreme reactivity and tendency of ROS to initiate and participate in chain reactions, the role of antioxidants as a defense system is highly recognized. Epidemiological studies revealed the link between reactive oxygen species, inflammation, ischemia and stroke risk. A key strategy to prevent potential damage to cellular compounds such as DNA, proteins and lipids is to reduce the free radical load [29].
The compounds were studied for their antioxidant activity by the use of the stable 2,2-diphenyl-1-picrylhydrazyl radical (DPPH) at concentration 0.1mM after 20 min. A freshly prepared DPPH solution exhibits a deep purple colour with an absorption maximum at 517 nm. This purple colour generally disappears in the presence of an antioxidant. The reduction of absorbance is a measure of the free DPPH due to the action of the antioxidant. The antioxidant activity was expressed as the RA% (Reducing Activity). The RA(%) values for the tested compounds of groups 4, 11, 13, 15 and 18 at 100µM, is very low, if any with the exception of compound 4j presenting 44% (Table 5) in comparison to the reference drug NDGA. Within the group of derivatives 4 the presence of the morpholinyl or piperidinyl rings at position A as well as the carbonyl group at position B, hinder the interaction of the compounds to the free stable radical DPPH. It seems that compound 4j with the combination of pyrrolidinyl ring and (CH2)2 chain (instead of a carbonyl group), present the structural features which support the interaction as well as the reducing ability and it does not face any stereochemical hindrance.
| Compds | RA% @ 100μΜ | ∙ΟΗ % @ 100μΜ | LP % @ 100μΜ | %LOX @ 100μΜ or IC50 μΜ | eeAChE % inhibn. @ 10 μΜ or IC50 μΜ | esBChE % inhibn. @ 10 μΜ or IC50 μΜ |
|---|---|---|---|---|---|---|
| 4a | nt | nt | 63 | 6% | nt | nt |
| 4b | nt | nt | no | 60 μΜ | 2.3 μΜ | 17% |
| 4c | 17 | no | 65 | no | nt | nt |
| 4d | nt | 59 | 21 | 44.5% | nt | nt |
| 4e | nt | nt | 65 | no | nt | nt |
| 4f | 3 | no | 62 | 53 μΜ | 1.4 μΜ | 36% |
| 4g | nt | nt | 16 | 10% | nt | nt |
| 4i | nt | nt | 54 | 2% | 7.7 μΜ | 15% |
| 4j | 44 | no | 32 | 37% | nt | nt |
| 4k | 12 | 27 | 57 | 10 μΜ | nt | nt |
| 4l | 12 | 90 | 37.5 | 61 μΜ | nt | nt |
| 4n | 17 | no | no | 35μΜ | nt | nt |
| 4o | 3 | no | 40 | 60 μΜ | nt | nt |
| 11a | nt | nt | 44 | no | nt | nt |
| 11b | 5 | no | no | no | 5.9 μΜ | 8% |
| 11d | 13 | no | 43 | 48μΜ | nt | nt |
| 11e | nt | nt | 63 | no | 7.8 μΜ | 8% |
| 11f | nt | nt | 48 | no | 43% | 14% |
| 11g | nt | nt | no | no | nt | nt |
| 13a | no | 90 | 100 | no | 40% | 23% |
| 13b | no | 95 | 90 | 44% | 3.1 μΜ | 13% |
| 13d | no | 79 | no | no | nt | nt |
| 13e | no | 99 | 100 | 62 μΜ | 1.73 μΜ | 18 μM |
| 13f | no | 100 | 23 | 25% | nt | nt |
| 13g | no | 100 | 43 | 62 μΜ | nt | nt |
| 13h | no | 97 | 86 | 55 μΜ | nt | nt |
| 13i | no | no | 74 | 76 μΜ | 5.0 μΜ | 18% |
| 15a | no | 93 | 100 | 55 μΜ | nt | nt |
| 15b | no | 48 | 52 | 100 μΜ | nt | nt |
| 15c | no | 97 | 43 | 9% | nt | nt |
| 18a | 4 | no | No | 90 μΜ | nt | nt |
| 18b | 1 | no | 38 | 100 μΜ | nt | nt |
| NDGA | 87 | 5.5 μΜ | ||||
| Trolox | 83 | 76 | ||||
| Galantamine | 0.51 μΜ | 8.7 μΜ |
Superoxide (O2-.) anion and hydroxyl radical (.OH) are free radical species of potential importance. In the acidic conditions of ischemic brain, O2-. is probably protonated to give HO2-. species. Iron released from damaged brain cells is more likely to be readily available to catalyze the generation of OH radicals. Among the ROS, the hydroxyl (•OH) free radical is possibly the most toxic, as it reacts with a number of biological important molecules. Polyunsaturated fatty acids are found in high concentrations in the CNS, and are particularly vulnerable by free radicals. Thus, we tried to test the ability of our compounds to scavenge hydroxyl radicals. The competition of compounds with DMSO for HO., generated by the Fe3+/ascorbic acid system, expressed as percent inhibition of formaldehyde production, was used for the evaluation of their hydroxyl radical scavenging activity In this experiment, the 13f, 13g, 13e, 13h, 15c, 15a, 13b, 13a and 4l showed remarkable activity at 100 μM, with values higher than the well known antioxidant trolox (Table 5). A number of compounds like 4c, 4f, 4j, 4n, 4o, 11b, 11d, 13i,18a, 18b did not present any activity whereas 4d, 4k, 13d and 15d showed lower response. Within the compounds 4a-4o the most potent derivatives 4d and 4j contain a morpholinyl ring in their structure which seems to be correlated with their scavenging activity. All the derivatives of series 13 (except of 13i) present high antioxidant activity (80-100%) which is not able to be correlated with any specific structural characteristic since all contain a coumarin, a purine and an isoxazolyl moiery. This observation is not followed within 15a-c where both 15a and c are almost equipotent whereas 15b the morpholinyl analogue exhibits half of their activity (48%). It seems that the overall molar configuration influences the response. However, antioxidants with hydrophilic or lipophilic character are both needed to act as radical scavengers in the aqueous phase or as chain-breaking antioxidants in biological membranes.
Anti-lipid peroxidation activity. The water-soluble azo compound AAPH has been extensively used as a clean and controllable source of thermally produced alkyl peroxyl free radicals, through spontaneous thermal decomposition. The use of the free radical reactions initiator AAPH is recommended as more appropriate for measuring radical-scavenging activity in vitro, because the activity of the peroxyl radicals produced by the action of AAPH shows a greater similarity to cellular activities such as lipid peroxidation. In the AAPH assay, the highly reactive alkylperoxyl radicals are intercepted mainly by a hydrogen atom transfer (HAT) from the antioxidant. Compounds 13a, 13b, 13e, 15a presented high activity whereas 4a, 4c, 4e, 4f, 11e, 13i, 13h showed 62-86% inhibition of lipid peroxidation. The rest exhibited limited or no activity (Table 5).
LO is the key enzyme in leukotriene biosynthesis [30]. Leukotrienes derived from the biotransformation of arachidonic acid catalyzed by 5-lipoxygenase (5-LO), are important inflammatory mediators [31] implicated in several diseases. LOs play a role in membrane lipid peroxidation by forming hydroperoxides in the lipid bilayer. Inhibitors of LO have attracted attention initially as potential agents for the treatment of inflammatory diseases. Most of the LO inhibitors are antioxidants or free radical scavengers, since lipoxygenation occurs via a carbon-centered radical [32, 33]. The evaluation of the novel coumarin hybrids against soybean lipoxygenase LO was accomplished by the UV-based enzyme assay [34].
Study of LO inhibition values demonstrates that compound 4k provided the best activity (IC50 = 10μM) followed by 4n (35μΜ), 11d (48μΜ), 4f (53μΜ), 13h and 15a (55μM), 4b (60mM), 4l (61mΜ), 13e and 13g (62.5μM), 13i (76μΜ), 18a (90μΜ), 15b and 18b (100μM). It seems that the ester 4k is more interesting and potent hybrid compared to the corresponding ether 4a. However, ester 4l is almost equipotent to ether 4b. Also the presence of a 4-methyl group enhances activity. Thus, 4k is more potent inhibitor compared to the 4n in which the methyl group is missing. The nature of A ring is a structural characteristic of importance. Thus, the piperidinyl derivative 4n is more potent (35μΜ) compared to 4o in which a morpholinyl group has replaced the piperidinyl group. The presence of pyrrolidinyl ring in compounds 4g (10%), 11g (no), 13g (62.5 μΜ) is correlated with no or low activity.
Within the compounds of 11a-g subgroup no inhibitory activities were observed. The only exception was compound 11d. Again a piperidinyl 6-substituted coumarin derivative was found to be more potent (IC50 = 48μM).
Among the isoxazole coumarin derivatives 13a-i most interesting results were given by 13h (55 μM), 13e (62.5μM), 13g (62.5 μM), and 13i (76 μM) (13h>13g, 13e>13i). The most potent 13h was a piperidinyl substituted derivative, whereas the replacement by a morpholinyl ring (13i) led to a decrease. Thus, the nature of the ring was implicated in the biological response. The equimolar response of 13e and 13i support the idea that the nature of A ring did not affect the inhibition.
Changing the attachment position of the isoxazole ring the analogues 15a-c were taken, from which 15a was more potent (55 μM) followed by 15b (100 μM) and 15c (9%). These findings follow the previous one supporting the significant role of the piperidinyl group. Also the piperidinyl derivative 18a was found slightly more active than the 18b.
Herein, the antilipid peroxidation activity does not go in parallel to the anti-LO activity (Table 5). Hydroxyl scavenging activity also was not found to be correlated with the above responses.
Considering the interesting results shown in Table 5 that clearly confirm the antioxidant potential of some of the new hybrid derivatives, we found interesting to test them as ChE inhibitors. Using already described protocols for the determination of the AChE [35] inhibition we obtained the IC50 values shown in Table 5. In Table 5 are only given the IC50 values for AChE inhibition of the more active compounds as well as the % inhibition for BChE. For comparative purposes some reference molecules have been incorporated.
Regarding the AChE inhibition (Table 5), hybrids 4f>13e>4b>13b>13i>11b>11e>4i showed significant eeAChE inhibition activity and they all are ethers of coumarin. The 4-, 6, or 7- position of attachment does not influence the result. For esBChE, most of the hybrids were poor inhibitors, with the exception of 13e, one of the two most potent AChE inhibitors (4f and 13e). All tested hybrids were less potent compared to the reference compound galantamine, but they retain fair AChE inhibitory activities in the low micromolar range.
For the MAO-A and B inhibition, we choose a subgroup consisted of the active anti-AChE compounds (Table 6). Only compound 11b showed an interesting inhibition activity IC50 = 9.5 μΜ against MAO-B acting as a selective agent. All the others present low (%) or not any activity at 10 μΜ. Hybrid 11b showed very lower inhibitory potency compared with the reference clorgyline, a well-established MAO-A selective inhibitor used in the treatment of depression. Indeed, the interesting MAO-B selectivity can be considered a good starting point for a structure-based refinement, in view of the potential of MAO-B selective inhibitors as neuroprotective agents in the therapy of neurodegenerative diseases [36].
| Compounds | MAO-A (%) @ 10μΜ | MAO-B (%) @ 10μΜ or IC50 μΜ |
|---|---|---|
| 4b | no | 24 |
| 4f | no | no |
| 4i | 25 | no |
| 11b | 15 | IC50 = 9.5 μΜ |
| 11e | no | 9 |
| 11f | 16 | 8 |
| 13a | 47 | 33 |
| 13b | 12 | no |
| 13e | 18 | 15 |
| 13i | 6 | 9 |
| Clorgyline | IC50 = 2.4 nM | IC50 = 2.4 μΜ |
Considering the MAO inhibition data, we conclude that hybrid 11b is a moderate, but selective inhibitor. Hybrids 4b, 4f, 4i, 11b, 11e, 13b, 13i are potent and selective inhibitors for AChE, whereas 13e is a dual AChE and BChE inhibitor.
CONCLUSION
In summary, the observed antioxidant activity of the majority of the examined hybrids, allows us to propose them as templates in the design of compounds useful in treating of AD that involves reactive oxygen species (ROS). Eleven out of twenty one derivatives are potent hydroxyl radical scavengers and significant number of them inhibit in vitro lipid peroxidation. Compounds 4k and 4n present higher LO inhibitory activity among the tested derivatives. It should to be noticed that compound 13e presents an antioxidant significant profile combining anti-LO, anti-AChE and anti-MAO-B activities. These results support the idea of a new lead compound. Overall the presented results would be possible to lead to a new multifunctional group of compounds.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
No Animals/Humans were used for studies that are base of this research.
CONSENT FOR PUBLICATION
Not applicable.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
The authors gratefully acknowledge Dr. A. Leo and Biobyte Corp. 201 West 4th Street, Suite 204, Claremont, CA 91711, USA for free access to the C-QSAR program.
SUPPORTIVE/SUPPLEMENTARY MATERIAL
The experimental data for all compounds are involved.
SUPPLEMENTARY MATERIAL
Supplementary material is available on the publishers Website along with the published article.
REFERENCES
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]

