All published articles of this journal are available on ScienceDirect.

Synthesis and Functional Evaluation of Novel Aldose Reductase Inhibitors Bearing a Spirobenzopyran Scaffold
Authors Info & Affiliations
Abstract
Background:
Aldose reductase, the first enzyme of the polyol pathway, is the key determinant for the pathogenesis of long term diabetic complications. Accordingly, its inhibition represents the major therapeutic strategy to treat this kind of pathologies.
Objectives:
In this work we describe the synthesis and the functional evaluation of a number of spiro-oxazolidinone and spiro-morpholinone acetic acid derivatives, and their benzyloxy analogs, developed as aldose reductase inhibitors.
Results:
Most of them proved to inhibit the target enzyme, showing IC50 values in the micromolar/low micromolar range. SARs observed among the three different series allowed to highlight their key pharmacophoric elements, thus creating sound basis for the design of novel and more effective inhibitors.
Conclusion:
Although further substitution patterns are needed, the novel compounds here proposed represent a good starting point for the development of novel and effective ARIs.
INTRODUCTION
Diabetes mellitus (DM) is one of the most common chronic metabolic disorders, characterized by elevated levels of blood glucose (hyperglycemia). According to “WHO Global report on diabetes”, the number of people with diabetes has risen from 108 million in 1980 to 422 million in 2014 [1]. The DM etiopathology is very complex and is closely related to the onset of chronic complications such as neuropathy, nephropathy, cataracts, retinopathy, accelerated atherosclerosis, and increased cardiovascular risk [2]. Therefore, nowadays, this disease is reasonably considered as a public health issue.
Many studies suggested the inhibition of aldose reductase (ARL2) enzyme as the key therapeutic strategy to prevent and/or ameliorate long-term diabetic complications [2]. ARL2 is the first enzyme of the polyol pathway; it is located in cytosol and it was found in eye lens, retina kidney, adrenal gland and some reproductive organs. ALR2 catalyzes the NADPH-dependent conversion of glucose to sorbitol, the rate-limiting step of the polyol pathway (Fig. 1) [3]; then the sorbitol produced is converted into fructose by sorbitol dehydrogenase (SDH). Since its hydrophilic nature, this sugar alcohol does not pass easily through cell membranes, thus accumulating within the cells while being slowly converted into fructose. The accumulation of sorbitol leads to osmotic stress in cells, event strongly related to the increasing of NAD+/NADH ratios. As a consequence, an impairment of cellular defense against oxidative injury occurs, leading to the onset of both micro- and macrovascular complications such as retinopathy, peripheral vascular disease and coronary artery disease. These events are frequently found in DM and are well known as “complications of secondary diabetes”. Moreover, many studies demonstrated an ALR2-dependent increase of ROS levels in cells cultured under high glucose conditions, thus confirming the leading role of this enzyme in the pathogenesis of many diabetic complications [4].
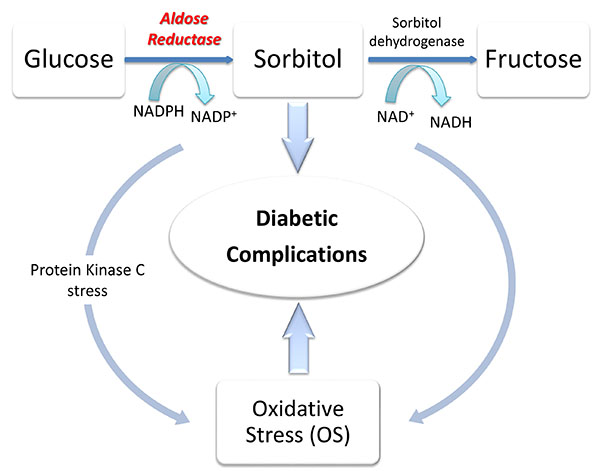
Therefore, ARL2 is considered an interesting target to counteract DM complications. In the last years, many ARL2 inhibitors (ARI) have been developed [5, 6] and some of them reached clinical trials to prove their efficacy in preventing and/or modulating/ameliorating DM complications. The most important ARIs can be classified into two chemical groups: (a) cyclic imides, such as sorbinil, fidarestat, minalrestat and ranirestat (AS-3201) [7] and (b) acetic acid derivatives, such as tolrestat, epalrestat, zopolrestat and ponalrestat [8]. To date, only the carboxylic acid epalrestat is available on the market and used for the treatment of diabetic neuropathy in Japan, India and China [9]. Most of the ARIs that showed to be active in in vitro tests failed the subsequent clinical trials, mainly because of their pharmacokinetic drawbacks and, consequently, low in vivo efficacy, besides the onset of adverse side effects. Notably, the side effects are principally due to the lack of selectivity of ARI toward aldehyde reductase (ALR1, EC 1.1.1.2). This last enzyme, ALR1, plays a detoxification role in specifically metabolizing toxic aldehydes such as hydroxynonenal (HNE), 3-deoxyglucosone, and methylglyoxal [10]. Zopolrestat, which belongs to the carboxylic acid derivatives, is quite selective for ALR2 vs ALR1 and other enzymes. However, the carboxylic class of agents becomes highly protein bound in vivo thus limiting their efficacy in vivo. On the other hand, ARIs bearing a hydantoin core, including sorbinil, are relatively non-selective and inhibit ALR1 and ALR2 with comparable efficacies.
Despite being chemically different, ARIs respond to some structural requirements represented by an acidic group, which interacts with the anionic site of the catalytic site, and a lipophilic group, binding the hydrophobic pocket, necessary for the activity and selectivity towards the enzyme [11].
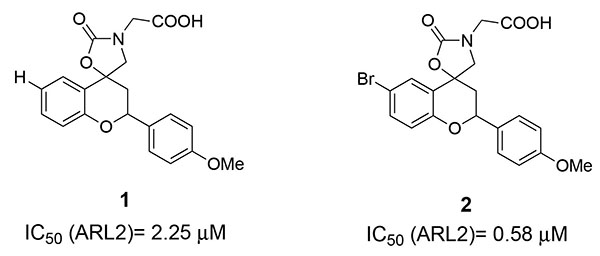
In a previous work we synthesized new spirobenzopyran derivatives (compounds type A, Fig. 2), as effective ARIs [12]. The previously synthesized ALR2 inhibitors possessed IC50 values in the submicromolar range and a good selectivity against the target enzyme.
Starting from these encouraging results, herein we performed a SAR study to investigate the effects on the ARL2 inhibitory activity induced by small structural modifications on the scaffold of type A compounds (Fig. 2).
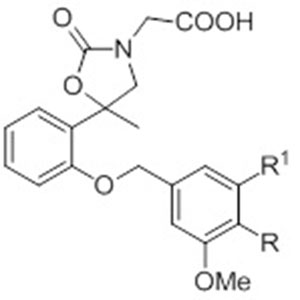
In particular, we synthesized the analogues with gem-dimethyl group in 2-position of the chromane-scaffold (5-10) to evaluate the influence of the steric hindrance in this position on the interaction with the active site of the enzyme. Then, in order to assess the importance of the benzopyran scaffold and the steric effects of substituents in the 2-position of the core, we synthesized compounds 11-13, in which the C2-C3 bond of the benzopyran nucleus has been cleaved, and compounds 5-10, in which the pendant phenyl ring on the benzopyran core has been replaced by gem-dimethyl groups.
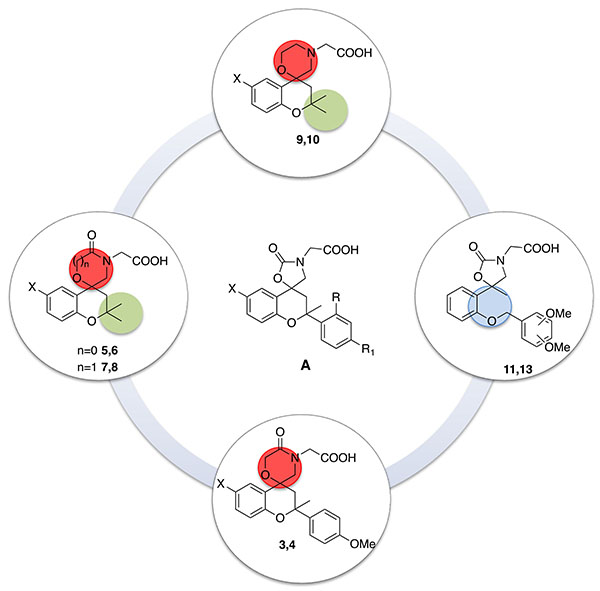
The new molecules synthesized were evaluated in vitro for their affinity and potency against the target enzyme, ARL2.
RESULTS AND DISCUSSION
Chemistry
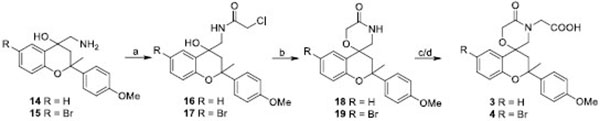
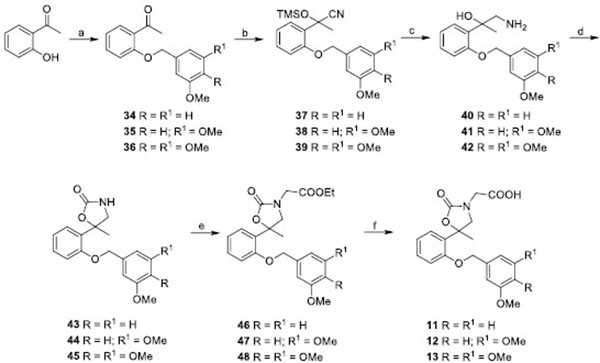
The acetic acid derivatives 3,4 have been synthesized as reported in Scheme 1. Briefly, the reaction of aminoalcohols14,15 [12] and chloroacetyl chloride in a heterogeneous phase yielded the chloroacetamides 16,17 which were submitted to a base-catalized (t-BuOK) cyclization to afford the spiromorpholone derivatives 18,19. The subsequent reaction of 18,19 with ethyl bromoacetate and NaH in DMF gave the corresponding esters, which were then cleaved in high yields to the carboxylic acids 3 and 4, respectively.
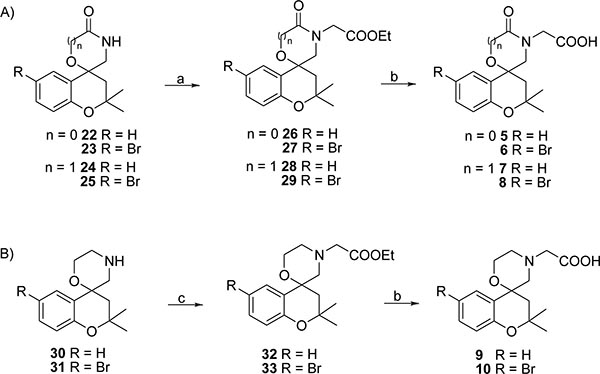
A tandem reaction of spiro-oxazolidinones 22,23 [13] and spiromorpholones 24,25 [14] with ethyl bromoacetate and the following cleavage of the intermediates (26-29), performed with KOH in MeOH, yielded the carboxylic acids 5-8 as the final products (Scheme 2A).
Spiromorpholine derivative 30 was obtained from 24 by reduction with LiAlH4. Notably, to avoid the possible halogen/metal exchange induced by LiAlH4, the 6-bromide-spiromorpholine 31 was obtained by a selective reduction of 25 [15] performed with a borane-methyl sulfide complex which can tolerate different substituents such as halogens or nitro-groups [16-19]. Subsequently, the reaction of products 30,31 with ethyl bromoacetate in the presence of K2CO3 gave compounds 32,33, which were then cleaved to the corresponding carboxylic acids 9,10 by the use of KOH/MeOH mixture (Scheme 2B).
Compounds 11-13 were synthesized starting from 2-hydroxyacetophenone and the appropriate benzyl chloride. Benzyloxyphenylethanones 34-36 were subjected to a nucleophilic addition with trimethylsilyl cyanide (TMSCN), in the presence of zinc iodide (ZnI2), to give the corresponding trimethylsilyl cyanohydrins 37-39. The reduction of 37-39, followed by a cyclization reaction with carbonyl diimidazole (CDI) gave the spirooxalidinone derivatives 43-45, which were then submitted to the same reaction of alkylation and cleavage above reported for compounds 9,10 to yield the final products as carboxylic acid 11-13 (Scheme 3).
Biological Evaluation
Recently, we developed a new class of small molecules bearing a benzopyran scaffold that displayed inhibitory activity against ARL2, with a high degree of selectivity when compared to the effects induced against ARL1 [12].
Among the previously studied derivatives, compounds 1 (IC50 2.25 μM) and 2 (IC50 0.58 μM), depicted in Fig. (3), shown the best results in terms of potency against the target enzyme.
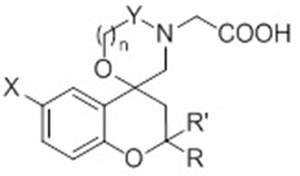
Previous studies highlighted that the ARL2 inhibition activity was influenced by the presence of both a bromine atom on the benzopyran scaffold and the substituent in the 2-position of the chromane nucleus. With the aim to extend the structure-activity relationship (SAR) study of this class of compounds, herein we describe the synthesis of new molecules, 3-13, in which a series of structural modifications have been performed as following reported: (a) replacement of the spirooxazolidine ring in the core structure of 1 and 2 with a spiromorpholone (derivatives 3 and 4) or with a spiromorpholine nucleus (derivatives 9 and 10); (b) replacement of the phenyl ring in the 2-position of the benzopyran scaffold with small lipophilic groups such as the methyl ones (derivatives 5-10) and (c) change in the conformational freedom of spirooxazolidine derivatives 1 and 2 through the cleavage of the C2-C3 bond within the benzopyran core (derivatives 11-13).
All the synthesized compounds were evaluated for their inhibitory activity against ARL2 and for their selectivity against aldehyde dehydrogenase (ALR1). Sorbinil, a well-known ARL2 inhibitor characterized by a spiro-chromane core, was used as the reference standard. Results obtained are reported in Tables 1 and 2, expressed as IC50 values.
 |
||||||
| Compounds | N | Y | X | R | R′ | IC50 (μM)a |
|---|---|---|---|---|---|---|
| Sorbinil | 0.65 | |||||
| 1b | 0 | CO | H | 4-OMe-Ph | H | 2.25 |
| 2b | 0 | CO | Br | 4-OMe-Ph | H | 0.58 |
| 3 | 1 | CO | H | 4-OMe-Ph | Me | n.a. |
| 4 | 1 | CO | Br | 4-OMe-Ph | Me | 4.97 |
| 5 | 0 | CO | H | Me | Me | 42.3 |
| 6 | 0 | CO | Br | Me | Me | 17.3 |
| 7 | 1 | CO | H | Me | Me | 2.00 |
| 8 | 1 | CO | Br | Me | Me | n.a. |
| 9 | 1 | CH2 | H | Me | Me | n.a. |
| 10 | 1 | CH2 | Br | Me | Me | 4.17 |
The synthesized compounds exhibited IC50 values in the micromolar/low micromolar range, with the only exception of compounds 3, 8 and 9 which proved to be completely inactive.
Noteworthy, results depicted in Table 1 show that the replacement of the spirooxazolidine nucleus of the leads, 1 and 2 [17], with the wider spiromorpholone core, like in compounds 3 and 4, induced a decrease of inhibitory potency against ARL2. Actually, compound 3, the higher homologue of derivative 1, turned out to be devoid of any inhibitory activity while compound 4, the higher homologue of 2, showed an almost 8-fold reduction in inhibitory efficacy when compared to the smaller parent (4, IC50 4.97 vs 2, IC50 0.58 μM).
 |
|||
| Compounds | R | R1 | IC50(μM)a |
|---|---|---|---|
| Sorbinil | 0.65 | ||
| 11 | H | H | 4.13 |
| 12 | H | OMe | nt |
| 13 | OMe | OMe | 3.19 |
We then decided to further investigate the effect of the substituent in the 2-position of the benzopyran core. In particular, we replaced the p-methoxyphenyl ring with a gem-substitution, in which two methyl groups were added. Regarding the spirooxazolidine series, this kind of chemical manipulation induced a significant decrease in the inhibitory potency of the resulting compounds, 5 and 6, even if the presence of a 6-bromine atom on the chromane core, like in 6, helped to keep the activity in the low micromolar range (IC50 17.3 μM). On the contrary, the same substitution pattern on the spiromorpholone series (derivatives 7 and 8) gave rise to contrasting results. Actually, while the gem-dimethyl substituents conferred a significant inhibitory activity to compound 7 (IC50 2.0 μM), the concomitant presence of a 6-bromine atom on the chromane core, like in 8, induced a dramatic loss of activity.
With the aim of reducing the conformational restriction of the spiromorpholone derivatives 7 and 8, we synthesized the spiromorpholine analogs, 9 and 10. This kind of structural modification was able to restore the inhibitory activity of the 6-bromine substituted 10 (IC50 =4.17μM). This result let us speculate that further improvement in the conformational freedom of this kind of compounds could enhance their inhibitory potency. On this basis, we replaced the benzopyran scaffold with the more flexible benzyloxy nucleus, thus synthesizing compounds 11-13.
As reported in Table 2, the novel compounds turned out to be almost 1.5-fold less potent that the parent 1, demonstrating that the cleavage of the C2-C3 bond of the previous synthesized compound 1 induced only a slight decrease of ARL2 inhibitory potency. Reasonably, the inhibitory activity of the novel series could be modulated by introducing suitable substituents on the benzyloxy nucleus. Accordingly, further investigation with both electron-withdrawing and electron-donating groups is needed in order to investigate their role in the identification of novel and more effective ARL2 inhibitors.
All the synthesized compounds, 3-13, were also tested for their ability to inhibit ALR1. Unlike the spiro-derivative sorbinil, that showed a significant inhibitory activity against ALR1 (IC50 0.029 μM), no inhibition was observed up to 100 μM of test compounds (data not shown), thus our compounds proved to be completely selective for the target ALR2.
MATERIALS AND METHODS
Chemistry
Melting points were determined on a Kofler hot-stage apparatus and are uncorrected. 1H NMR spectra were obtained with a Varian Gemini 200 MHz spectrometer whereas 13C NMR were obtained with a Bruker TopSpin 3.2 400 MHz spectrometer. Chemical shifts (δ) are reported in parts per million (ppm) downfield from tetramethylsilane and referenced from solvent references. The elemental compositions of the compounds agreed to within 0.4% of the calculated value. Chromatographic separation was performed on silica gel columns by flash (Kieselgel 40, 0.040–0.063 mm; Merck). Reactions were followed by thin layer chromatography (TLC) on Merck aluminum silica gel (60 F254) sheets that were visualized under a UV lamp. Evaporation was performed in vacuo (rotating evaporator). Sodium sulfate was always used as the drying agent. Commercially available chemicals were purchased from Sigma-Aldrich.
General Procedure for the Synthesis of Compounds 3-13
To a stirred solution of appropriate ester 20-21, 26-29, 32-33, 46-48 (0.77 mmol) in MeOH (3 mL) was added KOH 50% (0.005 mL) and the resulting mixture was refluxed for 2 h. The solvent was evaporated under vacuum and then HCl 1N was added (pH = 3). The precipitate was filtered and dried.
2-(2-(4-Methoxyphenyl)-2-methyl-5'-oxospiro[chroman-4,2'-morpholin]-4'-yl) acetic acid3
Compound 3 was obtained from 20 (0.47 g, 1.10 mmol) following procedure previously described without further purification. 3 (0.09 g, 0.23mmol, yield 21%). Mp175-178°C.1H-NMR (DMSO-d6): δ 1.63 (s, 3H, Me); 2.30 (d, 1H, J = 12.6 Hz, CH2); 2.42 (d, 1H, J = 14.3 Hz, CH2); 2.92 (d, 1H, J = 14.3 Hz, CH2); 3.25 (d, 1H, J = 17.2 Hz, CH2); 3.62 (d, 1H, J = 12.6 Hz, CH2); 3.70 (s, 3H, OMe); 4.16 (d, 1H, J = 17.2 Hz, CH2); 4.22 (d, 1H, J = 17.2 Hz, CH2); 4.35 (d, 1H, J = 17.2 Hz, CH2); 6.88 (d, 2H, J = 8.7 Hz, Ar); 6.94-7.04 (m, 2H, Ar); 7.26-7.42 (m, 4H, Ar) ppm. 13C NMR (400 MHz; DMSO-d6): δ 169.83, 166.13, 158.21, 153.34, 136.73, 130.18, 127.68, 125.98, 122.51, 120.68, 116.98, 113.80, 78.04, 68.51, 63.21, 55.30, 55.09, 48.12, 38.42, 29.33 ppm. Anal. (C22H23NO6) C, H, N. % Calcd: 66.49 (C); 5.83 (H); 3.52 (N). % Found: 66.65 (C); 6.00 (H); 3.63 (N).
2-(6-Bromo-2-(4-methoxyphenyl)-2-methyl-5'-oxospiro[chroman-4,2'-morpholin]-4'-yl) acetic acid4
Compound 4 was obtained from 21 (0.2 g, 0.4 mmol) following the procedure previously described. The crude product was purified by crystallization from (iPr)2O to give 4 (0.03 g, 0.07 mmol, yield 18%) 1HNMR (DMSO-d6): δ 1.64 (s, 3H, Me); 2.30-2.39 (m, 2H, CH2); 2.92 (d, 1H, J = 14.6 Hz, CH2); 3.25-3.38 (m, 2H, CH2); 3.54-3.65 (m, 1H, CH2); 3.72 (s, 3H, OMe); 4.08-4.40 (m, 2H, CH2); 6.89 (d, 2H, J = 8.8 Hz, Ar); 7.03 (d, 1H, J = 8.6 Hz, Ar); 7.31 (d, 2H, J = 8.8 Hz, Ar); 7.48 (dd, 1H, J = 2.4, 8.6 Hz, Ar); 7.55 (d, 1H, J = 2.4 Hz, Ar) ppm. Anal. (C22H22BrNO6) C, H, N. Anal. % Calcd: 55.47 (C); 4.66 (H); 2.94 (N). % Found: 55.52 (C); 4.66 (H); 2.94 (N).
2-(2,2-Dimethyl-2'-oxospiro[chroman-4,5'-oxazolidin]-3'-yl) acetic acid5
Compound 5 was obtained from 26 (0.23 g, 0.56 mmol) following procedure previously described without further purification. 5 (0.04 g, 0.16 mmol, yield 29%). Mp 160-165 °C. 1H-NMR (DMSO-d6): δ 1.32 (s, 3H, Me); 1.36 (s, 3H, Me); 2.27 (d, 1H, J = 14.8 Hz, CH2); 2.37 (d, 1H, J = 14.8 Hz, CH2); 3.68 (d, 1H, J = 9.1 Hz, CH2); 3.87 (d, 1H, J = 9.1 Hz, CH2); 3.95 (d, 1H, J = 17.7 Hz, CH2); 4.08 (d, 1H, J = 17.7 Hz, CH2); 6.80 (d, 1H, J = 8.7 Hz, Ar); 6.98 (m, 1H, Ar); 7.26 (m, 1H, Ar); 7.58 (d, 1H, J =8.7 Hz, Ar) ppm. Anal. (C15H17NO5) C, H, N. Anal. % Calcd: 61.85 (C); 5.88 (H); 4.81 (N). % Found: 61.60 (C); 5.85 (H); 4.79 (N).
2-(6-Bromo-2,2-dimethyl-2'-oxospiro[chroman-4,5'-oxazolidin]-3'-yl) acetic acid6
Compound 6 was obtained from 27 (0.33 g, 0.67 mmol) following procedure previously described without further purification. 6 (0.12 g, 0.35 mmol, 53%yield). Mp165-170°C. 1H-NMR (DMSO-d6): δ 1.32 (s, 3H, Me); 1.36 (s, 3H, Me); 2.27 (d, 1H, J = 15.0 Hz, CH2); 2.37 (d, 1H, J = 15.0 Hz, CH2); 3.68 (d, 1H, J = 9.2 Hz, CH2); 3.88 (d, 1H, J = 9.2 Hz, CH2); 3.90 (d, 1H, J = 18.1 Hz, CH2); 4.00 (d, 1H, J = 18.1 Hz, CH2); 6.79 (d, 1H, J = 8.7 Hz, Ar); 7.43 (dd, 1H, J = 2.3, 8.7 Hz, Ar); 7.85 (d, 1H, J = 2.3 Hz, Ar) ppm. Anal. (C15H16BrNO5) C, H, N. % Calcd: 48.67 (C); 4.36 (H); 3.78 (N). % Found: 48.3 (C); 4.70 (H); 3.50 (N).
2-(2,2-Dimethyl-5'-oxospiro[chroman-4,2'-morpholin]-4'-yl) acetic acid7
Compound 7 was obtained from 28 (0.26 g, 0.77 mmol) following procedure previously described. The crude product was collected without further purification to yield 7 (0.14 g, 0.46 mmol, 60% yield). Mp 163-165°C. 1H-NMR (CDCl3): δ 1.41 (s, 6H, Me); 2.19 (d, 1H, J = 14.6 Hz, CH2); 2.42 (d, 1H, J = 14.6 Hz, CH2); 3.26 (d, 1H, J = 12.1 Hz, CH2); 4.02- 4.10 (m, 2H, CH2); 4.35- 4.46 (m, 3H, CH2); 6.84- 7.0 (m, 2H, Ar); 7.22-7.29 (m, 1H, Ar); 7.46- 7.47 (m, 1H, Ar) ppm. 13C NMR (400 MHz; CDCl3): δ 171.88, 168.30, 153.95, 130.58, 127.53, 121.34, 120.95, 118.42, 74.46, 69.43, 63.42, 57.81, 48.10, 39.65, 26.19 ppm. Anal. (C16H19NO5) C, H, N. % Calcd: 62.94 (C); 6.27 (H); 4.59 (N). % Found: 62.40 (C); 5.83 (H); 4.09 (N).
2-(6-Bromo-2,2-dimethyl-5'-oxospiro[chroman-4,2'-morpholin]-4'-yl) acetic acid8
Compound 8 was obtained from 29 (0.39 g, 0.96 mmol) following the general procedure. The crude product was collected without further purification to yield 8 (0.12 g, 0.32 mmol, 34% yield). Mp 132-134°C. 1H-NMR (DMSO-d6): δ 1.32 (s, 3H, Me); 1.35 (s, 3H, Me); 2.14 (d, 1H, J = 15.0 Hz, CH2); 2.42- 2.52 (m, 1H, CH2); 3.96- 4.24 (m, 6H, CH2); 6.79 (d, 1H, J = 8.7 Hz, Ar); 7.40 (dd, 1H, J = 2.4, 8.7 Hz, Ar); 7.60 (d, 1H, J = 2.4 Hz, Ar) ppm. Anal. (C16H18BrNO5) C, H, N. % Calcd: 50.02 (C); 4.72 (H); 3.65 (N). % Found: 49.00 (C); 4.18 (H); 2.58 (N).
2-(2,2-Dimethylspiro[chroman-4,2'-morpholin]-4'-yl) acetic acid9
Compound 9 was obtained from 32 (0.55 g, 1.90 mmol) following procedure previously described. The crude product was collected and purified by precipitation from CHCl3/n-Hexane to yield 9 (0.01 g, 0.04 mmol, yield 2%). 1H-NMR (CD3OD-d4): δ 1.35 (s, 3H, Me); 1.45 (s, 3H, Me); 2.20 (d, 1H, J = 14.8 Hz, CH2); 2.65 (d, 1H, J = 14.8 Hz, CH2); 3.26-3.40 (m, 2H, CH2); 3.70-4.01 (m, 3H, CH2); 4.07-4.24 (m, 1H, CH2); 4.90 (s, 2H, CH2); 6.78- 6.83 (m, 1H, Ar); 6.93- 7.00 (m, 1H, Ar); 7.20- 7.28 (m, 1H, Ar); 7.55 (dd, 1H, J = 1.5, 7.9 Hz, Ar) ppm. Anal. (C16H21NO4) C, H, N. % Calcd: 65.96 (C); 7.27 (H); 4.81 (N). % Found: 65.69 (C); 7.24 (H); 4.79 (N).
2-(6-Bromo-2,2-dimethylspiro[chroman-4,2'-morpholin]-4'-yl) acetic acid10
Compound 10 was obtained from 33 (0.03 g, 0.11 mmol) and chloroacetic acid (0.01 g, 0.11 mmol) following the general procedure. The crude was used without further purification. 10 (0.005 g,0.07 mmol, yield 67%). 1H-NMR (CDCl3): δ 1.33 (s, 3H, Me); 1.40 (s, 3H, Me); 2.14 (d, 1H, J = 14.5 Hz, CH2); 2.54 (d, 1H, J = 14.5 Hz, CH2); 2.70-3.00(m, 4H, CH2); 3.19-3.42 (m, 2H, CH2); 3.78-3.84 (m, 1H, CH2); 3.99-4.10 (m,1H, CH2); 6.71 (d, 1H, J = 8.8 Hz, Ar); 7.29 (dd, 1H, J = 2.4, 8.8 Hz, Ar); 7.64(d, 1H, J = 2.4 Hz, Ar) ppm. Anal. (C16H20BrNO4) C, H, N. Anal. (C16H21NO4) C, H, N. % Calcd: 51.90 (C); 5.44 (H); 3.78 (N). % Found: 51.74 (C); 5.42 (H); 3.76 (N).
2-(5-(2-((3-Methoxybenzyl)oxy)phenyl)-5-methyl-2-oxooxazolidin-3-yl)acetic acid11
Compound 11 was obtained from 46 (0.05 g, 0.12 mmol) following procedure previously described. The crude product was collected and purified by precipitation from EtOAc/n-Hexane to yield 11 (0.02 g, 0.06 mmol, 49% yield). Mp117-119 °C. 1H-NMR (CDCl3): δ 1.75 (s, 3H, Me); 3.67-3.79 (m, 2H, CH2); 3.83 (s, 3H, OMe); 4.02-4.11 (m, 2H, CH2); 5.00 (s, 2H, CH2); 6.91-7.02 (m, 4H, Ar); 7.24-7.32 (m, 3H, Ar); 7.58 (dd, 1H, J = 1.8, 8.1 Hz, Ar) ppm. Anal. (C20H21NO6) C, H, N. % Calcd: 64.68 (C); 5.70 (H); 3.77 (N). % Found: 64.98 (C); 5.84 (H); 3.57 (N).
2-(5-(2-((3,5-Dimethoxybenzyl)oxy)phenyl)-5-methyl-2-oxooxazolidin-3-yl)acetic acid12
Compound 12 was obtained from 47 (0.05 g, 0.12 mmol) following same procedure described above. The crude was used without further purification. 12 (0.04 g, 0.11 mmol, 90% yield). Mp118-120 °C. 1H-NMR (CDCl3): δ 1.79 (s, 3H, Me); 3.65-3.90 (m, 5H, CH2, OMe); 4.03-4.17 (m, 2H, CH2); 4.94-5.08 (m, 2H, CH2); 6.83-7.04 (m, 5H, Ar); 7.26-7.33 (m, 1H, Ar); 7.59-7.63 (m, 1H, Ar) ppm. Anal. (C21H23NO7) C, H, N. % Calcd: 62.83 (C); 5.78 (H); 3.49 (N). % Found: 63.02 (C); 5.67 (H); 3.44 (N).
2-(5-Methyl-2-oxo-5-(2-((3,4,5-trimethoxybenzyl)oxy)phenyl)oxazolidin-3-yl)acetic acid13
Compound 13 was obtained from 48 (0.54g, 1.18 mmol) following procedure previously described. The crude product was dissolved in EtOAc and washed with NaHCO3 and then extracted with CH2Cl2. The organic layers were dried and concentrated under vacuum to give 13 (0.07 g, 0.15 mmol, 13% yield).1H-NMR (CDCl3): δ 1.83 (s, 3H, Me); 3.57-4.18 (m, 7H, CH2, OMe); 5.02 (s, 2H, CH2); 6.61 (s, 2H, Ar); 6.95-7.05 (m, 2H, Ar); 7.26-7.34 (m, 1H, Ar); 7.59-7.63 (m, 1H, Ar) ppm. 13C NMR (400 MHz; CDCl3): δ 171.92, 157.54, 154.16, 153.58, 137.83, 132.12, 131.37, 129.41, 125.79, 121.33, 112.05, 105.19, 80.66, 70.46, 61.03, 57.43, 56.27, 44.95, 27.15 ppm. Anal. (C22H25NO8) C, H, N. % Calcd: 61.25 (C); 5.84 (H); 3.25 (N). % Found: 61.06 (C); 5.82 (H); 3.24 (N).
General Procedure for Synthesis of Compounds 6-17
A solution of NaOH (0.19 g, 4.75 mmol) in H2O (8.5 mL) was added to a solution appropriate amine 14-15 (4.00 mmol) [12] in CH2Cl2 (12 mL). The mixture was stirred, cooled to 0° C and treated dropwise with chloroacetyl chloride (0.73 g, 6.5 mmol). At the end of the addiction, the mixture was stirred vigorously at r.t. for 1 h. The layers were separated and the CH2Cl2 solution was washed with diluted HCl and H2O, dried and evaporated.
2-Chloro-N-((4-hydroxy-2-(4-methoxyphenyl)-2-methylchroman-4-yl)methyl)acetamide16
Compound 16 was obtained from 14 (1.6 g, 5.35 mmol) and chloroacetyl chloride (0.86 g, 7.49 mmol) following the general procedure. The crude product was directly used in the next step without further purification. 16 (1.30 g, 3.47 mmol, yield 65%).1H-NMR (CDCl3): δ 1.67 (s, 3H, Me); 2.29 (d, 1H, J = 14.2 Hz, CH2); 2.63 (d, 1H, J = 14.2 Hz, CH2); 3.45 (dd, 1H, J = 5.8, 14.4 Hz, CH2); 3.74 (d, 1H, J = 14.4 Hz, CH2); 3.75 (s, 3H, OMe); 3.99 (d, 1H, J = 15.5 Hz, CH2); 4.09 (d, 1H, J = 15.5 Hz, CH2); 6.79-6.90 (m, 3H, Ar); 6.94-7.02 (m, 1H, Ar); 7.18-7.36 (m, 3H, Ar); 7.45 (d, 1H, J = 7.6 Hz, Ar) ppm.
N-((6-Bromo-4-hydroxy-2-(4-methoxyphenyl)-2-methylchroman-4-yl)methyl)-2-chloroacetamide17
Compound 17 was obtained from 15 (0.8 g, 2.11 mmol) and chloroacetyl chloride (0.34 g, 2.95 mmol) following the general procedure. The crude product was directly used in the next step without further purification. 17 (0.8 g, 1.76 mmol, yield 47%). 1H-NMR (CDCl3): δ 1.69 (s, 3H, Me); 2.24-2.36 (m, 2H, CH2); 2.66 (d, 1H, J = 14.2 Hz, CH2); 3.38-3.48 (m, 1H, CH2); 3.78 (s, 3H, OMe); 4.03 (d, 1H, J = 15.7 Hz, CH2); 4.13 (d, 1H, J = 15.7 Hz, CH2); 6.80-6.94 (m, 3H, Ar); 7.24-7.38 (m, 3H, Ar); 7.61 (d, 1H, J = 2.6 Hz, Ar) ppm.
General Procedure for Synthesis of Compounds18-19
Potassium tert-butoxide (1.45 g, 13 mmol) was added portion wise over 1 h to a stirred solution of appropriate alcohol 16-17 (2.5 mmol) in toluene (20 mL). The reaction mixture was stirred at r.t. for 2 h, then the solvent was evaporated. The residue was diluted with EtOAc and the organic layer washed with water, dried and evaporated.
2-(4-Methoxyphenyl)-2-methylspiro[chroman-4,2'-morpholin]-5'-one18
Compound 18 was obtained from 16 (1.30 g, 3.47 mmol) following the procedure previously described. The crude product was directly used in the next step without further purification. 18 (0.73 g, 2.16 mmol, yield 63%). 1H-NMR (CDCl3): δ 1.72 (s, 3H, Me); 2.24 (d, 1H, J = 12.8 Hz, CH2); 2.33 (d, 1H, J = 13.7 Hz, CH2); 3.01 (d, 1H, J = 13.7 Hz, CH2); 3.33 (d, 1H, J = 12.8 Hz, CH2); 3.76 (s, 3H, OMe); 4.38 (s, 2H, CH2); 6.80 (d, 2H, J = 7.6 Hz, Ar); 6.92-6.99 (m, 1H, Ar); 7.03-7.07 (m, 1H, Ar); 7.15-7.39 (m, 4H, Ar) ppm.
6-Bromo-2-(4-methoxyphenyl)-2-methylspiro[chroman-4,2'-morpholin]-5'-one19
Compound 19 was obtained from 17 (0.8 g, 1.76 mmol) following the procedure previously described. The crude product was directly used in the next step without further purification. 19 (0.63 g, 1.5 mmol, yield 85%). 1H-NMR (CDCl3): δ 1.74 (s, 3H, Me); 2.29 (d, 1H, J = 12.8 Hz, CH2); 2.35 (d, 1H, J = 13.7 Hz, CH2); 3.05 (d, 1H, J = 13.7 Hz, CH2); 3.40 (d, 1H, J = 12.8 Hz, CH2); 3.78 (s, 3H, OMe); 4.40 (s, 2H, CH2); 6.82 (d, 2H, J = 8.8 Hz, Ar); 7.03 (d, 1H, J = 8.6 Hz, Ar); 7.29-7.35 (m, 2H, Ar); 7.45-7.50 (m, 1H, Ar); 7.55 (d, 1H, J = 2.4 Hz, Ar) ppm.
General Procedure for Synthesis of Compounds 20-21, 26-29
To a stirred solution of NaH (0.07 g, 3.00 mmol, 60% dispersion in mineral oil) in dry DMF (10 mL) was added the appropriate derivatives 18-19, 22-25 (4.00 mmol) under N2 atmosphere. After 30 min, the reaction mixture was cooled at 0 °C and ethyl bromoacetate (0.83 g, 5.00 mmol) was added. The reaction mixture was allowed to warm at 25 °C and stirred for 1h before being quenched with water and extracted with EtOAc. The combined organic layers were dried, filtered, and concentrated under vacuum.
Ethyl 2-(2-(4-methoxyphenyl)-2-methyl-5'-oxospiro[chroman-4,2'-morpholin]-4'-yl) acetate20
Compound 20 was obtained from 18 (0.73 g, 2.16 mmol) and ethyl bromoacetate (0.78 g, 2.16 mmol) following the general procedure. The crude product was purified by flash column chromatography eluting n-Hexane/EtOAc (1:1) to give 20 (0.47 g, 1.10 mmol, yield 51%): 1H-NMR (CDCl3): δ 1.26 (t, 3H, J = 7.1 Hz, Me); 1.73 (s, 3H, Me); 2.21 (d, 1H, J = 12.6 Hz, CH2); 2.39 (d, 1H, J = 14.2 Hz, CH2); 3.04 (d, 1H, J = 14.2 Hz, CH2); 3.18 (d, 1H, J = 17.3 Hz, CH2); 3.64 (d, 1H, J = 12.6 Hz, CH2); 3.78 (s, 3H, OMe); 4.16 (q, 2H, J = 7.1 Hz, CH2); 4.34 (d, 1H, J = 17.3 Hz, CH2); 4.41 (s, 2H, CH2); 6.82 (d, 2H, J = 8.7 Hz, Ar); 6.93-7.08 (m, 2H, Ar); 7.21-7.36 (m, 3H, Ar); 7.44 (dd, 1H, J = 8.7, 1.4 Hz, Ar) ppm.
Ethyl 2-(6-bromo-2-(4-methoxyphenyl)-2-methyl-5'-oxospiro[chroman-4,2'-morpholin]-4'-yl) acetate21
Compound 21 was obtained from 19 (0.3 g, 0.72 mmol) and ethyl bromoacetate (0.12 g, 0.72 mmol) following the general procedure. The crude product was purified by flash column chromatography eluting n-Hexane/EtOAc (1:1) to give 21 (0.22 g, 0.4 mmol, yield 55%). 1H-NMR (CDCl3): δ 1.30 (t, 3H, J = 7.0 Hz, Me); 1.75 (s, 3H, Me); 2.23 (d, 1H, J = 12.4 Hz, CH2); 2.40 (d, 1H, J = 13.2 Hz, CH2); 3.01 (d, 1H, J = 13.2 Hz, CH2); 3.15 (d, 1H, J = 17.0 Hz, CH2); 3.61 (d, 1H, J = 12.4 Hz, CH2); 3.78 (s, 3H, OMe); 4.20 (q, 2H, J = 7.0 Hz, CH2); 4.30 (d, 1H, J = 17.0 Hz, CH2); 4.35 (s, 2H, CH2); 6.72 (d, 2H, J = 8.6 Hz, Ar); 6.97 (d, 1H, J = 8.3 Hz, Ar); 7.00 (d, 2H, J = 8.6 Hz, Ar); 7.60 (dd, 1H, J = 2.5, 8.3 Hz, Ar); 7.65 (d, 1H, J = 2.5 Hz, Ar) ppm.
Ethyl 2-(2,2-dimethyl-2'-oxospiro[chroman-4,5'-oxazolidin]-3'-yl) acetate26
Compound 26 was obtained from 22(0.44 g, 1.37 mmol) following procedure previously described. The crude product was directly used for the next reaction without further purification. 26 (0.23 g, 0.56 mmol, yield 41%): 1H-NMR (CDCl3): δ 1.33 (t, 3H, J =7.1 Hz, Me); 1.42 (s, 6H, Me); 2.17 (d, 1H, J =14.6 Hz, CH2); 2.46 (d, 1H, J =14.6 Hz, CH2); 3.64 (d, 1H, J =8.4 Hz, CH2); 3.98 (d, 1H, J =8.4 Hz, CH2); 4.02 (d, 1H, J =18.0 Hz, CH2); 4.20 (d, 1H, J =18.0 Hz, CH2); 4.25 (q, 2H, J =7.1 Hz, CH2); 6.82 (d, 1H, J =8.2 Hz, Ar); 6.94-7.02 (m, 1H, Ar); 7.20-7.29 (m, 1H, Ar); 7.58 (dd, 1H, J =8.0 Hz, Ar) ppm.
Ethyl 2-(6-bromo-2,2-dimethyl-2'-oxospiro[chroman-4,5'-oxazolidin]-3'-yl) acetate27
Compound 27 was obtained from 23 (0.55 g, 1.37 mmol) following procedure previously described. The crude product was directly used for the next reaction without further purification. 27 (0.33 g, 0.67 mmol, yield 49%): 1H-NMR (CDCl3): δ 1.32 (t, 3H, J = 7.1 Hz, Me); 1.41 (s, 6H, Me); 2.16 (d, 1H, J = 14.7 Hz, CH2); 2.45 (d, 1H, J = 14.7 Hz, CH2); 3.67 (d, 1H, J = 8.7 Hz, CH2); 3.93 (d, 1H, J = 8.7 Hz, CH2); 4.01-4.33 (m, 4H, CH2); 6.70 (d, 1H, J = 8.7 Hz, Ar); 7.33 (dd, 1H, J= 2.4, 8.7 Hz, Ar); 7.68 (d, 1H, J = 2.4 Hz, Ar) ppm.
Ethyl 2-(2,2-dimethyl-5'-oxospiro[chroman-4,2'-morpholin]-4'-yl) acetate28
Compound 28 was obtained from 24 (0.6 g, 2.42 mmol) and ethyl bromo acetate (0.48 g, 2.90 mmol) following the procedure previously described. The crude product was purified by flash column chromatography eluting n-Hexane/EtOAc (7:3) to give 27 (0.26 g, 0.77 mmol, yield 32%): 1H-NMR (CDCl3): δ 1.21- 1.41 (m, 9H, Me); 2.21 (d, 1H, J = 14.7 Hz, CH2); 2.42 (d, 1H, J = 14.7 Hz, CH2); 3.22 (d, 1H, J = 12.1 Hz, CH2); 3.93- 4.41 (m, 7H, CH2); 6.84 (dd, 1H, J = 1.4; 8.0 Hz, Ar); 6.90- 6.98 (m, 1H, Ar); 7.19- 7.28 (m, 1H, Ar); 7.49 (dd, 1H, J = 1.4; 8.0 Hz, Ar) ppm.
Ethyl 2-(6-bromo-2,2-dimethyl-5'-oxospiro[chroman-4,2'-morpholin]-4'-yl) acetate29
Compound 29 was obtained from 25 (0.33 g, 1.00 mmol) and ethyl bromo acetate (0.20 g, 1.20 mmol) following the procedure previously described. The crude product was purified by flash column chromatography eluting n-Hexane/EtOAc (7:3) to give 29 (0.18 g, 0.44 mmol, yield 44%) 1H-NMR (CDCl3): δ 1.20-1.41 (m, 9H, Me); 2.20-2.45 (m, 2H CH2); 3.20 (d, 1H, J = 12.2 Hz, CH2); 3.90- 4.44 (m, 7H, CH2); 6.73 (d, 1H, J = 8.7 Hz, Ar); 7.32 (dd, 1H, J = 2.5, 8.7 Hz, Ar); 7.61 (d, 1H, J= 2.5 Hz, Ar) ppm.
General Procedure for Synthesis of Compounds 32-33
To a solution of appropriate derivatives 30-31 (0.57 mmol) in MeCN (5 mL) was added K2CO3 (0.09 g, 0.64mmol) and ethyl bromoacetate (0.09 g, 5.00 mmol). The resulting mixture was refluxed for 12 h, then, after cooling, was filtered and the solvent evaporated.
Ethyl 2-(2,2-dimethylspiro[chroman-4,2'-morpholin]-4'-yl) acetate32
Compound 32 was obtained from 30 (0.45 g, 1.92 mmol) and ethyl bromoacetate (0.32 g, 1.92 mmol) following the same procedure described in the general procedure. The crude product was used for the next step without further purification. 32 (0.55 g, 1.90 mmol, yield 99%): 1H-NMR (CDCl3): δ 1.22-1.33 (m, 3H, Me); 1.36 (s, 3H, Me); 1.41 (s, 3H, Me); 2.28 (d, 1H, J = 14.8 Hz, CH2); 2.56 (d, 1H, J = 14.8 Hz, CH2); 2.65-2.90 (m, 2H, CH2); 3.19 (d, 1H, J = 16.7 Hz, CH2); 3.37 (d, 1H, J = 16.7 Hz, CH2); 3.73-3.82 (m, 3H, CH2); 4.01-4.28 (m, 3H, CH2); 6.78-6.96 (m, 2H, Ar); 7.14-7.23 (m, 1H, Ar); 7.56 (dd, 1H, J = 1.5, 7.8 Hz, Ar) ppm.
Ethyl 2-(6-bromo-2,2-dimethylspiro[chroman-4,2'-morpholin]-4'-yl) acetate33
Compound 33 was obtained from 31 (0.6 g, 2.42 mmol) and ethyl bromoacetate (0.40 g, 2.42 mmol) following the procedure previously described. The crude product was purified by flash column chromatography eluting n-Hexane/ EtOAc (7:3) to give 33.(0.56 g, 1.98 mmol, yield 97%): 1H-NMR (CDCl3): δ 1.20- 1.30 (m, 3H, Me); 1.33 (s, 3H, Me); 1.39 (s, 3H, Me); 2.25 (d, 1H, J = 14.5 Hz, CH2); 2.56 (d, 1H, J = 14.5 Hz, CH2); 2.67 (d, 1H, J = 10.0 Hz, CH2); 2.93 (d, 1H, J = 10.0 Hz, CH2); 3.19 (d, 1H, J = 16.4 Hz, CH2); 3.37 (d, 1H, J = 16.4 Hz, CH2); 3.69-3.80 (m, 3H, CH2); 3.99-4.11 (m, 3H, CH2); 6.79 (d, 1H, J = 8.7 Hz, Ar); 7.4 (dd, 1H, J = 2.5, 8.7 Hz, Ar); 7.6 (d, 1H, J = 2.5 Hz, Ar) ppm.
General Procedure for Synthesis of Compounds 34-36
To a solution of 2-hydroxyacetophenone (1.5 g, 11.03 mmol) in DMSO (7 mL) was added KOH (1.86 g, 33.09 mmol) and the resulting mixture was stirred at 50°C for 15 min. After cooling to r.t. the appropriate methoxybenzyl chloride (11.03 mmol) was added. The reaction mixture was stirred at r.t. for 4h and then washed with H2O and NaHCO3sat. The organic layer was dried, filtered and evaporated under vacuum.
1-(2-((3-Methoxybenzyl)oxy)phenyl)ethanone34
Compound 34 was synthesized from 2-hydroxyacetophenone (1.5 g, 11.03 mmol) and 4-methoxybenzyl chloride (1.73 g, 11.03 mmol) following the procedure previously described. The crude product was directly used in the next step without further purification. 34(1.5 g, 5.83 mmol, yield 53%).1H-NMR (CDCl3): δ 2.57 (s, 3H, Me); 3.83 (s, 3H, OMe); 5.09 (s, 2H, CH2); 6.93 (d, 2H, J = 8.6 Hz, Ar); 6.96-7.05 (m, 2H, Ar); 7.36 (d, 2H, J= 8.6 Hz, Ar); 7.40-7.49 (m, 1H, Ar); 7.75 (dd, 1H, J = 2.0, 7.7, Hz, Ar) ppm.
1-(2-((3,5-Dimethoxybenzyl)oxy)phenyl)ethanone35
Compound 35 was synthesised from 2-hydroxyacetophenone (1.5 g, 11.03 mmol) and 3,4-dimethoxybenzyl chloride (2.06 g, 11.03 mmol) following the same procedure described in general procedure. The crude product was directly used for the next reaction without further purification. 35 (1.83 g, 9.81 mmol, yield 89%): 1H-NMR (CDCl3): δ 2.59 (s, 3H, Me); 3.89 (s, 3H, OMe); 3.90 (s, 3H, OMe); 5.09 (s, 2H, CH2); 6.85-7.05 (m, 5H, Ar); 7.41-7.49 (m, 1H, Ar); 7.74 (dd, 1H, J =1.8, 7.5 Hz, Ar) ppm.
1-(2-((3,4,5-Trimethoxybenzyl)oxy)phenyl)ethanone36
Compound 36 was synthesised from 2-hydroxyacetophenone (0.9 g, 6.61 mmol) and 3,4,5-trimethoxybenzyl chloride (1.5 g, 6.93 mmol) following the same procedure previously described. The crude product was directly used for the next reaction without further purification. 36 (1.9 g, 6.03 mmol, yield 87%). 1HNMR (CDCl3): δ 2.63 (s, 3H, Me); 3.86 (s, 9H, OMe); 5.09 (s, 2H, CH2); 6.67 (s, 2H, Ar); 6.99-7.06 (m, 2H, Ar); 7.41-7.50 (m, 1H, Ar); 7.74 (dd, 1H, J = 1.7, 8.0 Hz, Ar) ppm.
General Procedure for Synthesis of Compounds 37-39
To a solution of appropriate derivatives 37-39 (1.23 g, 7 mmol) in CH2Cl2 was added trimethylsilylcyanide (TMSCN) (1.4 mL, 10.5 mmol) and ZnI2 (0.33 g, 1.05 mmol). The mixture was stirred at r.t. for 4 h, then CH2Cl2 was added and the solution was washed with water. The organic layer was dried and evaporated.
3-(2-((3-Methoxybenzyl)oxy)phenyl)-3-((trimethylsilyl)oxy)butanenitrile37
Compound 37 was synthesised from 34 (1.5 g, 5.83 mmol) following the procedure described in general procedure. The crude product was directly used in the next step without further purification. 37 (1.74 g, 4.88 mmol, yield 84%).1HNMR (CDCl3): δ 0.31 (s, 9H, Me); 1.89 (s, 3H, Me); 3.82 (s, 3H, OMe); 5.05-5.16 (m, 2H, CH2); 6.77-7.04 (m, 4H, Ar); 7.19-7.35 (m, 1H, Ar); 7.46 (d, 2H, J= 8.8 Hz; Ar); 7.56 (dd, 1H, J = 2.0, 8.8 Hz, Ar) ppm.
3-(2-((3,5-Dimethoxybenzyl)oxy)phenyl)-3-((trimethylsilyl)oxy)butanenitrile38
Compound 38 was synthesised from 35 (1.09 g, 5.83 mmol) following the procedure previously described. The crude product was directly used in the next step without further purification. 38 (1.91 g, 4.95 mmol, yield 85%): 1H-NMR (CDCl3): δ 0.29 (s, 9H, Me); 1.89 (s, 3H, Me); 3.89 (s, 3H, OMe); 3.90 (s, 3H, OMe); 5.03-5.18 (m, 2H, CH2); 6.67-7.03 (m, 5H, Ar); 7.18-7.41 (m, 1H, Ar); 7.56 (dd, 1H, J =1.8, 8.1 Hz) ppm.
3-(2-((3,4,5-Trimethoxybenzyl)oxy)phenyl)-3-((trimethylsilyl)oxy)butanenitrile39
Compound 39 was synthesised from 36 (1.90 g, 6.03 mmol) following the same procedure described in general procedure. The crude product was directly used in the next step without further purification. 39 (2.22 g, 5.37 mmol, yield 89%).1H-NMR (CDCl3): δ 0.26 (s, 9H, Me); 2.01 (s, 3H, Me); 3.88 (s, 9H, OMe); 5.18 (s, 2H, CH2); 6.80 (s, 2H, Ar); 7.01-7.08 (m, 2H, Ar); 7.35-7.42 (m, 2H, Ar) ppm.
General Procedure for Synthesis of Compounds 40-42
A solution of appropriate trimethylsilyl cyanohydrins 37-39 (5.00 mmol) in THF was added dropwise at 0°C to a solution of LiAlH4 (1M in THF, 10 mmol). The reaction mixture was stirred at r.t. for 1h, then quenched with water and NaOH. The resulting lithium salts were filtered and the solution was evaporated.
1-Amino-2-(2-((3-methoxybenzyl)oxy)phenyl)propan-2-ol40
Compound 40 was synthesised from 37 (1.74 g, 4.88 mmol) following the procedure described in the general procedure. The crude product was directly used in the next step without further purification. 40 (0.83 g, 2.88 mmol, yield 59%).1H-NMR (CDCl3): δ 1.49 (s, 3H, Me); 3.31 (d, 1H, J = 12.7 Hz, CH2); 3.51 (d, 1H, J = 12.7 Hz, CH2); 3.83 (s, 3H, OMe); 5.00-5.14 (m, 2H, CH2); 6.87-7.05 (m, 4H, Ar); 7.18-7.28 (m, 1H, Ar); 7.35 (d, 2H, J = 8.6 Hz, Ar); 7.68 (d, 1H, J = 2.0, 8.6Hz, Ar) ppm.
1-Amino-2-(2-((3,5-dimethoxybenzyl)oxy)phenyl)propan-2-ol41
Compound 41 was synthesised from 38 (1.91 g, 4.95 mmol) following the procedure described above. The crude product was directly used in the next step without further purification. 41 (1.19 g, 3.76 mmol, yield 76%).1H-NMR (CDCl3): δ 1.54 (s, 3H, Me); 2.77 (d, 1H, J =12.7 Hz, CH2); 3.29 (d, 1H, J =12.7 Hz, CH2); 3.88 (s, 3H, OMe); 3.89 (s, 3H, OMe); 5.03 (s, 2H, CH2); 6.62-7.02 (m, 5H, Ar); 7.20-7.28 (m, 1H, Ar); 7.45-7.50 (m, 1H, Ar) ppm.
1-Amino-2-(2-((3,4,5-trimethoxybenzyl)oxy)phenyl)propan-2-ol42
Compound 42 was synthesised from 39 (1.91 g, 4.95 mmol) following the procedure previously described. The crude product was directly used in the next step without further purification. 42 (1.21 g, 3.49 mmol, yield 65%) 1H-NMR (CDCl3): δ 1.57 (s, 3H, Me); 2.80 (d, 1H, J =12.7 Hz, CH2); 3.31 (d, 1H, J=12.7 Hz, CH2); 3.85 (s, 9H, OMe); 5.03 (s, 2H, CH2); 6.65 (s, 2H, Ar); 6.90-7.02 (m, 2H, Ar); 7.18-7.28 (m, 1H, Ar); 7.48-7.53 (m, 1H, Ar) ppm.
General Procedure for Synthesis of Compounds 43-45
A solution of aminoalcohols 40-42(6.33 mmol) in THF was added dropwise to a solution of N,N’-carbonyl diimidazole (CDI) (1.02 g, 6.33 mmol) in THF at 0 °C. The reaction mixtures were stirred r.t. for 5h. The solvent was evaporated under vacuum and the residue diluted with EtOAc and washed with HCl 1N and K2CO3sat. The organic layers were dried and concentrated under vacuum.
5-(2-((3-Methoxybenzyl)oxy)phenyl)-5-methyloxazolidin-2-one43
Compound 43 was synthesised from 40 (0.48 g, 1.67 mmol) following the general procedure. The crude product was purified by flash column chromatography eluted by n-Hexane/EtOAc (5.5:4.5) to give 43 (0.16 g, 0.5 mmol, yield 30%): 1H-NMR (CDCl3): δ1.73 (s, 3H, Me); 3.62 (d, 1H, J = 9.1 Hz, CH2); 3.72 (d, 1H, J = 9.1 Hz, CH2); 3.83 (s, 3H, OMe); 4.99 (s, 2H, CH2); 6.93 (d, 2H, J = 8.4 Hz, Ar); 6.96-7.04 (m, 2H, Ar); 7.25-7.33 (m, 3H, Ar); 7.59-7.63 (m, 1H, Ar) ppm.
5-(2-((3,5-Dimethoxybenzyl)oxy)phenyl)-5-methyloxazolidin-2-one44
Compound 44 was synthesised from 41 (0.53 g, 1.67 mmol) following the procedure described above. The crude product was directly used in the next step without further purification. 44 (0.45 g, 1.30 mmol, yield 78%).1H-NMR (CDCl3) δ: 1.75 (s, 3H, Me); 3.63 (d, 1H, J = 9.1 Hz, CH2); 3.73 (d, 1H, J = 9.1 Hz, CH2); 3.89 (s, 3H, OMe); 3.90 (s, 3H, OMe); 4.92-5.09 (m, 2H, CH2); 6.70-7.04 (m, 5H, Ar); 7.25-7.33 (m, 1H, Ar); 7.58-7.63 (m, 1H, Ar) ppm.
5-Methyl-5-(2-((3,4,5-trimethoxybenzyl)oxy)phenyl)oxazolidin-2-one45
Compound 45 was synthesised from 42 (1.21 g, 3.49 mmol) following the general procedure. The crude product was directly used in the next step without further purification. 45 (1.07 g, 2.86 mmol, yield 82%).1H-NMR (CDCl3): δ 1.80 (s, 3H, Me); 3.67-3.93 (m, 11H, CH2, OMe); 4.92-5.45 (m, 2H, CH2); 6.60 (s, 2H, Ar); 6.92-7.06 (m, 2H, Ar); 7.20-7.30 (m, 1H, Ar); 7.62 (dd, 1H, J = 1.6, 7.7 Hz, Ar) ppm.
General Procedure for Synthesis of Compounds 46-48
To a stirred solution of NaH (0.07 g, 3.00 mmol, 60% dispersion in mineral oil) in dry DMF (10 mL) was added appropriate compounds 43-45 (0.9 g, 4.00 mmol) under N2 atmosphere. After 30 min, the reaction mixture was cooled at 0 °C and ethyl bromoacetate (0.83 g, 5.00 mmol) was added. The reaction mixture was allowed to warm at 25 °C and stirred for 1 h before being quenched with water and extracted with EtOAc. The combined organic layers were dried, filtered, and concentrated under vacuum.
Ethyl 2-(5-(2-((3-methoxybenzyl)oxy)phenyl)-5-methyl-2-oxooxazolidin-3-yl)acetate46
Compound 46 was synthesised from 43 (0.16 g, 0.5 mmol) and ethylbromoacetate (0.08 g, 0.5 mmol) following the procedure described above. The crude product was directly used in the next step without further purification. 46 (0.16 g, 0.4 mmol, yield 81%): 1H-NMR (CDCl3): δ 1.18-1.27 (m, 3H, Me); 1.77 (s, 3H, Me); 3.66-3.78 (m, 2H, CH2); 3.83 (s, 3H, OMe); 4.02-4.26 (m, 4H, CH2); 5.01 (s, 2H, CH2); 6.91-7.04 (m, 4H, Ar); 7.26-7.33 (m, 3H, Ar); 7.59-7.63 (m, 1H, Ar) ppm.
Ethyl 2-(5-(2-((3,5-dimethoxybenzyl)oxy)phenyl)-5-methyl-2-oxooxazolidin-3-yl)acetate47
Compound 47 was synthesised from 44 (0.17 g, 0.5 mmol) and ethyl bromoacetate (0.08 g, 0.5 mmol) following the general procedure. The crude product was directly used in the next step without further purification. 47 (0.17 g, 0.4 mmol, yield 81%).1H-NMR (CDCl3): δ 1.14-1.33 (m, 3H, Me); 1.79 (s, 3H, Me); 3.65-3.98 (m, 10H, CH2, OMe); 4.02-4.25 (m, 2H, CH2); 5.01 (s, 2H, CH2); 6.70-6.79 (m, 1H, Ar); 6.83-7.03 (m, 4H, Ar); 7.24-7.33 (m, 1H, Ar); 7.58-7.62 (m, 1H, Ar) ppm.
Ethyl 2-(5-methyl-2-oxo-5-(2-((3,4,5-trimethoxybenzyl)oxy)phenyl)oxazolidin-3-yl)acetate48
Compound 48 was synthesised from 45 (0.50 g, 1.34 mmol) and ethyl bromoacetate (0.22 g, 1.34 mmol) following the procedure previously described. The crude product was directly used in the next step without further purification. 48 (0.54 g, 1.18 mmol, yield 88%).1H-NMR (CDCl3): δ 1.14-1.33 (m, 3H, Me); 1.84 (s, 3H, Me); 3.70-3.92 (m, 13H, CH2, OMe); 4.06-4.22 (m, 2H, CH2); 4.94-5.17 (m, 2H, CH2); 6.61 (s, 2H, Ar); 6.93-7.05 (m, 2H, Ar); 7.20-7.30 (m, 1H, Ar); 7.63 (dd, 1H, J = 1.6, 7.7 Hz, Ar) ppm.
Biology
Materials and Methods
Adult Sprague-Dawley albino rats (body weights of 120−140), provided by Harlan Nossan, Italy, were exploited to obtain the target enzymes, ALR2 and ALR1, which were isolated and purified from lens and kidney, respectively, following a previously reported protocol [17-19]. Pyridine coenzyme, D,L-glyceraldehyde, and sodium D-glucuronate came from Sigma-Aldrich while Sorbinil was a gift from Pfizer, Groton CT. All other chemicals were of reagent grade.
Enzymatic Assays
The activity of the two enzymes was determined spectrophotometrically by monitoring the change in absorbance at 340 nm, which is due to the oxidation of NADPH catalyzed by ALR2 and ALR1. The change in pyridine coenzyme concentration/min was determined using a Beckman DU-64 kinetics software program (Solf Pack TM Module).
ALR2 activity was determined at 30°C in a reaction mixture containing 10 mM D,L-glyceraldehyde, 0.104 mM NADPH, 0.1 M sodium phosphate buffer (pH 6.2), deionized water and the enzyme extract, in a total volume of 1 mL.
ALR1 activity was assayed at 37°C in a reaction mixture containing 20 mM sodium D-glucuronate, 0.12 mM NADPH, 0.1 M sodium phosphate buffer (pH 7.2) and the enzyme extract, in a total volume of 1 mL.
Enzymatic Inhibition
The new synthesized compounds were tested for the inhibitory activity against ALR2 and ALR1 by adding 0.1 mL of the inhibitor solution to the reaction mixture described above. The inhibitors were solubilized in water, and the pH was adjusted to 7. A reference blank containing all the above assay components except the substrate was prepared, to correct for the non-enzymatic oxidation of NADPH and for absorption by the compounds tested.
The inhibitory effect of the new derivatives was routinely estimated at a concentration of 10-4 M. Those compounds found to be active were tested at additional concentrations between 10-5 and 10-7 M. The IC50 values were determined through linear regression analysis of the log-dose response curve, generated using at least four concentrations of the inhibitor causing an inhibition between 20% and 80%, with two replicates at each concentration.
CONCLUSION
In this study, we described the design, synthesis and biological evaluation of three small series of spirooxazolidinone, spiromorpholinone and benzyloxy acetic acid derivatives, proposed as novel ALR2 inhibitors.
Moving from our previously developed spirochromanes 1 and 2 [12], and through suitable chemical manipulation of both their substitution pattern and their main heterocyclic structure, we succeeded in obtaining a novel set of analogues which proved to inhibit the target enzyme showing IC50 values in the micromolar/low micromole range.
According to SARs observed, the presence of a pendant phenyl ring in the 2-position of the central core is essential to confer a good inhibitory activity to the spirooxazolidine and spiromorpholine derivatives bearing a 6-bromine atom.
On the contrary, a gem-dimethyl substitution proves to be more fruitful in the case of the 6-unsubstituted analogues. The flexible benzyloxy derivatives benefit from the presence of methoxy groups in suitable positions of the pendant phenyl ring.
Although further substitution patterns are needed to extend and deepen SARs observed for the three sub-series, the novel compounds here proposed represent a good starting point for the obtainment of novel and effective ARIs.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
Declared none.
REFERENCES
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]

