All published articles of this journal are available on ScienceDirect.

Molecular Docking-based Screening of Natural Heterocyclic Compounds as a Potential Drug for COVID-19
Abstract
Introduction:
The coronavirus pandemic poses significant challenges for the pharmaceutical industry.
Methods:
Coronavirus enters host cells via the angiotensin-converting enzyme 2 receptors (ACE2).
The SARS-CoV-2 spike glycoprotein is a potential target for medicinal chemists in the development of specific drugs. The current study investigates molecular modeling studies to identify potential drug candidates. Molecular docking simulations were run on 11 natural heterocyclic compounds/flavonoids.
Results:
When tested against the viral spike protein receptor, isoquercetin had a docking binding energy of -6.74kcal/mol (PDBID:6LU7).
Conclusion:
A docking study revealed the interaction of the receptor-binding domain with various flavonoid compounds.
1. INTRODUCTION
Flavonoids are naturally occurring compounds found in fruits and vegetables that exhibit a variety of beneficial physiological activities [1]. Quercetin has three important medicinal properties: anti-inflammatory, antioxidant, and immunomodulatory. Due to combination of these three properties, quercetin is an excellent candidate for dealing with situations involving oxidative stress, inflammation, and the immune system [1]. The lower oral bioavailability of quercetin limits its clinical use [2]. In silico study of selected flavonoids aids in the identification of potential drug candidates. The purpose of this study is to investigate the anti-covid activity of some selected flavonoids using molecular docking analysis and assess the efficiency of quercetin in Covid-19 infection prevention.
Important pharmacological effects of quercetin include its antiviral, pro-metabolic, and anti-inflammatory properties. Moreover, a variety of anticancer qualities have been shown for it, and numerous publications support its effectiveness as a cancer preventative. The substance has a wide range of therapeutic benefits, including effects on the immune system, cardioprotection, and neuroprotection, as well as antioxidant and anti-inflammatory characteristics. The antioxidant and free radical scavenging properties of quercetin are primarily responsible for its neuroprotective benefits [3]. Flavonoids with strong binding to SARS-CoV-2 targets include apigenin and quercetin. Orally, isoquercetin is bioavailable. Isoquercetin functions as an antioxidant by scavenging free radicals and preventing oxidative cell damage [4].
Angiotensin-converting enzyme ACE2 is a membrane-associated protein found in a variety of cell types, including lung alveolar epithelial cells and vascular endothelial cells. SARS-coronaviruses, including the 2019-nCoV new coronavirus, enter the body through ACE2 [5].
It is possible to prevent this virus from entering cells if these receptors are not present.
Quercetin and Isoquercetin have often been at the top of the list of potential effectors in silico screening of small molecules for binding affinity to proteins involved in the SARS-CoV-2 life cycle. Clinical trials may be conducted to examine the preventative and therapeutic effects of these flavonoids for COVID-19 if these predictions hold true and in vitro tests confirm them [6, 7].
The best method for evaluating ligands against prospective therapeutic targets is in silico studies. In addition, lower binding energy is significant.
2. MATERIALS AND METHODS
2.1. Databases and Software Used
Pubchem, PDB, Molinspiration and Docking web servers were used for the study. Flavonoid compounds used for the study were taken from PubChem in SMILES (Simplified Molecular Input Line Entry System) format. Virtual screening of the compounds was carried out using Molecular Docking Webserver [8]
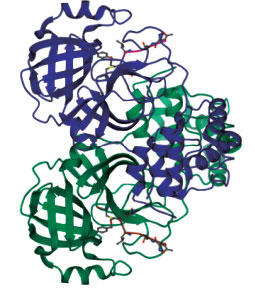
2.2. Preparation of Ligand and Protein
The PDB was used to retrieve the three-dimensional structures of the coronavirus's essential proteins [9] (PDBID: 6LU7). The receptor for this investigation, the major protease 6Lu7 of COVID-19, is shown in Fig. (1). Crystal structure. Apigenin, Chrystin, Diosmetin, Fisetin, Flavone, Hispidulin, Kaempferol, Luteolin, Quercetin, Recoflavone, and Isoquercetin were the 11 flavonoids employed in the study. Fig. (2) presents the two-dimensional structures derived from PubChem. The three-dimensional structures of the proteins and the ligand were loaded into the molecular docking web server, which uses Auto Dock 4.2. The active sites identified were provided in Auto Dock, the docking grid was set accordingly, and the docking was performed.
The docking protocol used in the docking webserver was used to dock the selected 11 compounds. The resultant binding energy values of these 11 compounds studied are presented in Table 2.
| S.No | Flavanoids | Smiles |
|---|---|---|
| 1 | Apigenin | C1=CC(=CC=C1C2=CC(=O)C3=C(C=C(C=C3O2)O)O)O |
| 2 | Chrysin | C1=CC=C(C=C1)C2=CC(=O)C3=C(C=C(C=C3O2)O)O |
| 3 | Diosmetin | COC1=C(C=C(C=C1)C2=CC(=O)C3=C(C=C(C=C3O2)O)O)O |
| 4 | Fisetin | C1=CC(=C(C=C1C2=C(C(=O)C3=C(O2)C=C(C=C3)O)O)O)O |
| 5 | Flavone | C1=CC=C(C=C1)C2=CC(=O)C3=CC=CC=C3O2 |
| 6 | Hispidulin | COC1=C(C2=C(C=C1O)OC(=CC2=O)C3=CC=C(C=C3)O)O |
| 7 | Kaempferol | C1=CC(=CC=C1C2=C(C(=O)C3=C(C=C(C=C3O2)O)O)O)O |
| 8 | Luteolin | C1=CC(=C(C=C1C2=CC(=O)C3=C(C=C(C=C3O2)O)O)O)O |
| 9 | Quercetin | C1=CC(=C(C=C1C2=C(C(=O)C3=C(C=C(C=C3O2)O)O)O)O)O |
| 10 | Recoflavone | COC1=C(C=C(C=C1)C2=CC(=O)C3=C(O2)C=C(C=C3OC)OCC(=O)O)OC |
| 11 | Isoquercetin | C1=CC(=C(C=C1C2=C(C(=O)C3=C(C=C(C=C3O2)O)O)OC4C(C(C(C(O4)CO)O)O)O)O)O |
|
Flavanoid Compounds |
Est. Free Energy of Binding (kcal/mol) |
Est. Inhibition Constant, Ki(uM) |
vdW + Hbond + desolv Energy (kcal/mol) |
Electrostatic Energy (kcal/mol) |
Total Intermolec. Energy (kcal/mol) |
Interact. Surface |
|---|---|---|---|---|---|---|
| Diosmetin | -5.21 | 151.27 | -5.27 | -0.07 | -5.34 | 502.259 |
| Fiestin | -5.22 | 149.14 | -5.50 | +0.22 | -5.28 | 566.088 |
| Flavone | -6.12 | 32.59 | -6.41 | -0.01 | -6.42 | 569.41 |
| Hispidulin | -4.86 | 275.59 | -5.04 | -0.03 | -5.07 | 469.854 |
| Quercetin | -5.52 | 89.67 | -5.03 | -0.08 | -5.11 | 516.389 |
| Apigenin | -4.23 | 797.00 | -5.68 | -0.02 | -5.70 | 620.061 |
| Chrysin | -5.48 | 96.59 | -5.62 | -0.07 | -5.69 | 499.882 |
| Isoquercetin | -6.74 | 11.51 | -4.86 | -0.08 | -4.94 | 524.747 |
| Kaemperol | -5.05 | 200.15 | -5.30 | -0.10 | -5.41 | 508.22 |
| Luteolin | -5.64 | 72.96 | -5.16 | -0.11 | -5.27 | 505.346 |
| Recoflavone | -3.40 | 3.23 mM | -5.71 | +0.20 | -5.50 | 667.845 |
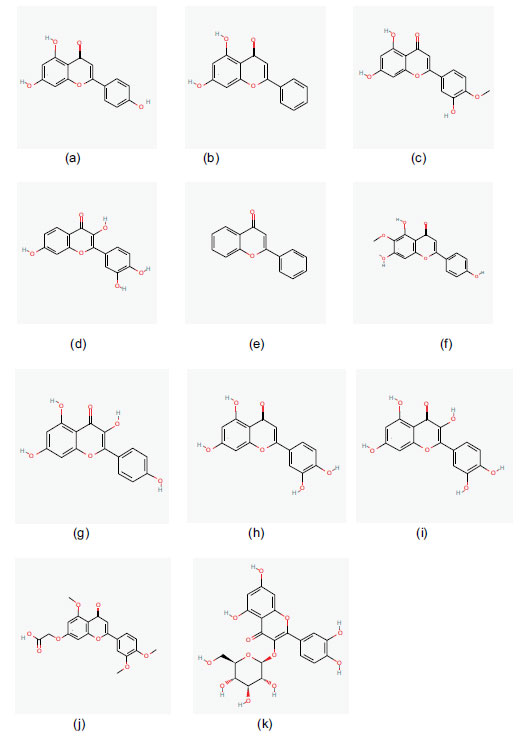
For molecular docking investigations, 11 flavonoids were specifically chosen. A strong interaction is indicated by the ligand and receptor's lower binding energies. In the current study, docking and molecular modeling are used to test 11 quercetin analogs. Significant inhibition was observed in the investigation, with binding energies ranging from -6.7 kcal/mol to -3.4 kcal/mol.
Depending on the specific outcomes of molecular docking, quercetin might have a potential impact on SARS-CoV-2. Proteases are crucial for the replication of viruses, and it has been revealed that the major protease in SARS-CoV-2 is 6LU7. The 6LU7 amino acids His164, Glu166, Asp187, Gln192, and Thr190 formed H-bonds with quercetin, all of which interacted with amino acids in the viral protease active site.
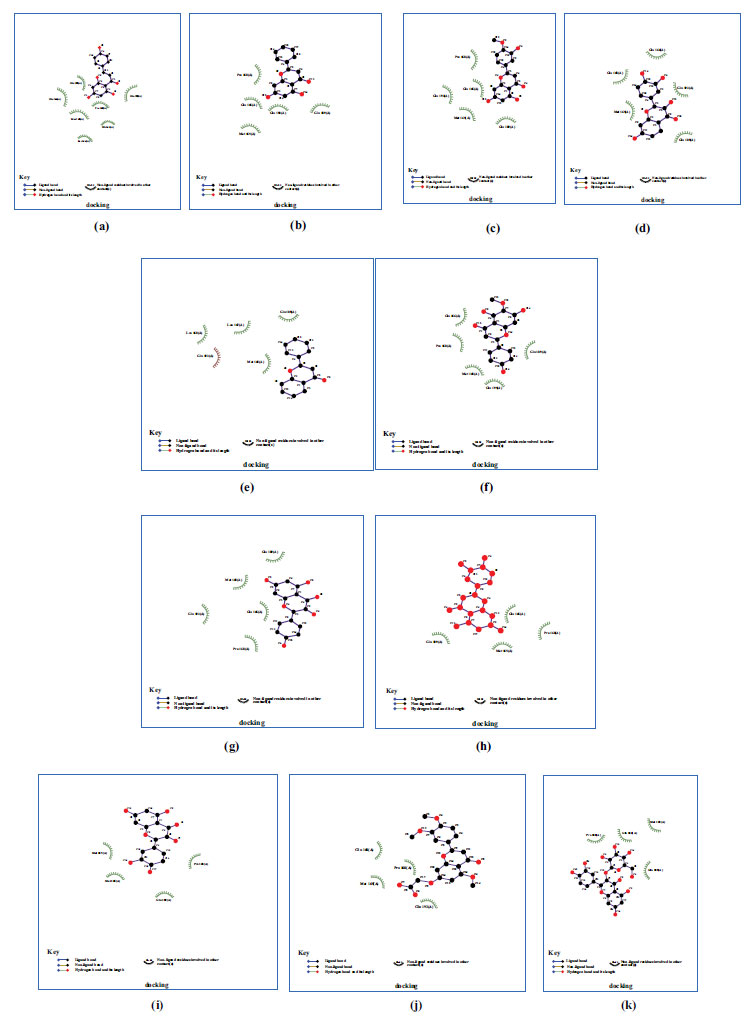
a) Apegenin b) Chrysin c) Diosmetin
d) Fisetin e) Flavone f) Hispidulin
g) Isoquercetin h) Kemperol i) Luteolin
j) Quercetin k) Recoflavone
2.3. Molecular Docking
Molecular docking was done on the compounds to investigate the binding interactions shown in Figs. (3 and 4) for the 11 compounds that were chosen. Table 1 displays the 11 compounds' respective binding energy values. The chemicals' binding conformations were carefully chosen based on both significant non-bonded interactions and binding energy levels. In addition, to calculate the favourable binding of ligand molecules, the free energy of binding, inhibitory constant (Ki), the total energy of vdW +disolv Energy, electrostatic energy, total inter-molecular energy, and interact surface were analysed and are shown in Table 2.
Gasteiger partial charges were added to the ligand atoms. Non-polar hydrogen atoms were merged, and rotatable bonds were defined. Docking calculations were carried out on the protein model. Essential hydrogen atoms, Kollman united atom type charges, and solvation parameters were added with the aid of AutoDock tools [9, 10]. Affinity (grid) maps of grid points and 0.375 Å spacing were generated using the Auto grid program. AutoDock parameter set- and distance-dependent dielectric functions were used in the calculation of the van der Waals and the electrostatic terms, respectively. Docking simulations were performed using the Lamarckian genetic algorithm (LGA) and the Solis & Wets local search method [11]. The initial position, orientation, and torsions of the ligand molecules were set randomly. All rotatable torsions were released during docking. Each docking experiment was derived from 10 different runs that were set to terminate after a maximum of 250000 energy evaluations. The population size was set to 150. During the search, a translational step of 0.2 Å and quaternion and torsion steps of 5 were applied.
Based on the study, all the compounds were found to have binding energies between -6.7 and -3.4 kcal/mol, indicating that most of the interaction process is exothermic.

| - | Polar | Hydrophobic | Other |
|---|---|---|---|
| Diosmetin | GLN189 (-1.3463) | PRO168 (-1.3247) | |
| GLU166 (-0.6105) | MET165 (-0.8349) | ||
| GLN192 (-0.4555) | - | ||
| Fiestin | GLN189 (-1.201) | PRO168 (-0.9556) | GLU166 (-2.1372) |
| GLN192 (-0.2713) | MET165 (-0.7584) | - | |
| Flavone | MET165 (-0.953) | GLN189 (-1.6921) | |
| LEU167 (-0.6282) | GLN192 (0.4844) | ||
| PRO168 (-0.2381) | - | ||
| Hispidulin | GLN192 (-0.3847) | PRO168 (-1.9374) | GLN189 (-1.0159) |
| GLU166 (-0.3381) | - | MET165 (-0.4254) | |
| Quercetin | GLU166 (-1.1314) | PRO168 (-1.5546) | MET165 (-0.6301) |
| GLN189 (-1.1193) | - | - | |
| Apigenin | GLN189 (-1.6085) | MET165 (-0.9788) | GLU166 (-1.2925) |
| GLN192 (-0.5173) | - | PRO168 (-0.4735) | |
| HIS163 (-0.333) | - | - | |
| SER144 (-0.2036) | - | - | |
| Chrysin | GLN189 (-1.2186) | PRO168 (-0.9679) | GLU166 (-0.1977) |
| GLN192 (-0.456) | MET165 (-0.8269) | - | |
| Isoquercetin | GLN189 (-1.365) | PRO168 (-1.6592) | |
| GLU166 (-1.0775) | MET165 (-0.6888) | ||
| Kaemperol | GLU166 (-0.8445) | PRO168 (-1.8989) | GLN189 (-1.3255) |
| GLN192 (-0.4619) | - | MET165 (-0.4731) | |
| Luteloin | GLN189 (-0.8833) | PRO168 (-1.5998) | MET165 (-0.6825) |
| - | GLU166 (-0.1076) | ||
| Recoflavone | MET165 (-0.6193) | - | GLU166 (-9.8421) |
| PRO168 (-0.3572) | GLN192 (-0.3432) |
The substances under study exhibited inhibitory activity, which indicates how well they succeeded in inhibiting the enzyme. The compound Isoquercetin had the lowest binding energy and the lowest inhibitory concentration (-6.74 Kcal/mol). Table 3's decomposed interaction energies revealed that the predominant interactions for flavone and isoquercitrin were hydrophobic and polar, respectively. The studied ligands showed a strong correlation between binding energy and the inhibition constant M.
3. RESULTS AND DISCUSSION
Table 2 displays the findings of the docking investigation. The ligands under study have binding energies and inhibition factors that range from -6.74 kcal/mol to -3.4 kcal/mol and 11.51 uM to 3.23 mM, respectively. Therefore, isoquercetin displayed the lowest binding energy and inhibition coefficient among the substances examined. The compounds' comparable binding sites demonstrate that functional groups were responsible for the binding properties.
The energy decomposition results for the selected 11 compounds in Table 3 revealed that the residues Met165, Pro168, Glu 166, Gln189, and Gln 192 play important roles in compound binding. Polar and hydrophobic interactions dominated interactions with these residues (Table 3). Quercetin demonstrated good binding affinity in a study by Zang et al. [12] that screened 115 compounds found in Chinese medicinal plants. Manjesh et al. [13] conducted a docking study in which they docked various coronavirus proteins with quercetin ligands and found binding energies ranging from -3.37kcal/mol to -6.71kcal/mol. Keretsu et al. used a variety of modeling experiments to identify protease inhibitors from a database of prospective 3CLpro inhibitors [14].
An open-label randomised controlled clinical trial conducted at King Edward Medical University's Department of Medicine under clinical trial id NCT04861298 suggests a therapeutic role for quercetin in early-stage COVID-19, including SARS-CoV-2 clearance, early resolution of acute symptoms, and modulation of the host's hyperinflammatory response [15]. Quercetin has the potential to treat severe inflammation and life-threatening conditions in COVID-19 patients [16]. According to the current study, isoquercetin has a higher binding affinity to the receptor than quercetin. Animal studies on oral bioavailability show that isoquercetin has higher oral bioavailability than quercetin [17].
In vitro studies of flavonoid compounds showed inhibitory activity against SARS CoV 2 Mpro IC50 values ranging from 0.125 to 12.9µM. Studies on the Inhibitory effects of quercetin and isoquercitrin on the replication of HCoV-229E in Huh-7 cells showed that Isoquercetin strongly inhibited the replication starting with 2.5 μM [18]. Quercetin is one of the most common flavonoid compounds having five hydroxyl groups with electron-donating activity at positions 3, 5, 7, 3’, and 4’ [19]. In addition, to the above isoquercetin bears a single glucose moiety while losing an OH group. This alteration not only increases its hydrophilicity and water solubility by four times compared to quercetin’s. After oral administration, isoquercetin produces a maximum plasma concentration of quercetin that is 1.7 to 10 times greater when pharmacokinetic characteristics are compared [20].
CONCLUSION
ACE2 is essential to the pathogenesis of SARS-CoV-2 because it acts as a receptor and a conduit for virus particles to enter, causing the recently discovered outbreak [21]. Due to its inhibitory effect on the expression of the human ACE2 receptors, SARSCoV-2 enzymes (MPro, PLPro, and RdRp), antioxidant activity, anti-inflammatory activity, and immunomodulatory activity, quercetin has the potential to be used as a preventative or therapeutic agent against COVID-19, as shown by clinical trials, inventive compositions, and patent literature [22].
Out of the 11 compounds we studied, all exhibited a good, expected affinity for the major protease of SARS-CoV-2, a top target in the COVID-19 drug discovery. Among the compounds screened, Isoquercetin displayed binding energy scores of -6.7 kcal/mol, which indicates a robust interaction with the protease target. Isoquercetin is a better drug than quercetin because it has lower binding energy for docking studies and higher oral bioavailability. The study recommends Isoquercetin as a potential molecule candidate for an anti-COVID-19 role. It strongly supports the execution of a case-control clinical study to realize its possible efficacy within the context of this disease.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
No animals/humans were used in this research.
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
The data and supportive information are available within the article.
FUNDING
None.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
Declared none.

