All published articles of this journal are available on ScienceDirect.

Fusion of Morpholine and Schiff Base on Novel Benzimidazole Scaffold as Anti-microbial Agents: A Computational Approach and In-vitro Evaluation
Authors Info & Affiliations
Abstract
Background
Benzimidazole is a well-known bioactive heterocyclic compound with diverse pharmacological properties. In this study, the fusion of morpholine and Schiff base motifs with benzimidazole has been explored to enhance their antimicrobial activity against bacterial and fungal pathogens. Computational methods complement the experimental findings by providing insights into binding interactions and drug-likeness properties.
Aims and Objective
The study aimed to synthesize novel benzimidazole derivatives fused with morpholine and Schiff basesand evaluate their antimicrobial efficacy and drug-likeness properties through experimental assays and computational modeling.
Methods
The titled compounds were synthesized following a multi-step chemical process and characterized by using various spectroscopic techniques. The antimicrobial activity of synthesized motifs was assessed against bacterial and fungal strains using in-vitro assays. Computational docking was performed to evaluate binding affinities with target enzymes, also pharmacokinetic and physicochemical properties were analyzed to determine drug-likeness properties.
Results
Synthesized derivatives demonstrated significant antimicrobial activity, particularly compounds 3b and 3e, which showed potent inhibition of bacterial and fungal pathogens. Computational studies confirmed favorable binding interactions and drug-likeness profiles, correlating well with in-vitro findings.
Conclusion
The study highlights the potential of benzimidazole derivatives fused with morpholine and Schiff bases as promising antimicrobial agents. These findings pave the way for further exploration of their therapeutic applications, particularly in combating antimicrobial resistance.
1. INTRODUCTION
Over the past decade, the emergence of infectious diseases has driven the development of numerous antimicrobial agents. Another factor is the development of anti-microbial resistance (AMR), which significantly complicates the fight against infections. Additionally, comorbidities, mortalities, and the high cost of healthcare products are making the current scenario the most serious and overwhelming [1, 2]. Nevertheless, the emergence of antibiotic resistance, a phenomenon that is expedited by the use of antibiotics [3, 4], has caused many previously effective antibiotics, antifungals, and other treatments to become ineffective due to the appearance of resistant microorganisms [5]. Tragically, it is projected that Asia will witness a staggering 4.7 million fatalities directly caused by antimicrobial resistance (AMR) by the year 2050 [6-8].
Hence, the development of novel antimicrobial agents is essential, and their preparation must consider their chemical behavior in comparison to existing drugs. With the help of well-known heterocycles, the most important part of drug development and strategy design in the synthesis of lead drugs is making sure that they are structurally very similar to their biological counterparts [9, 10].
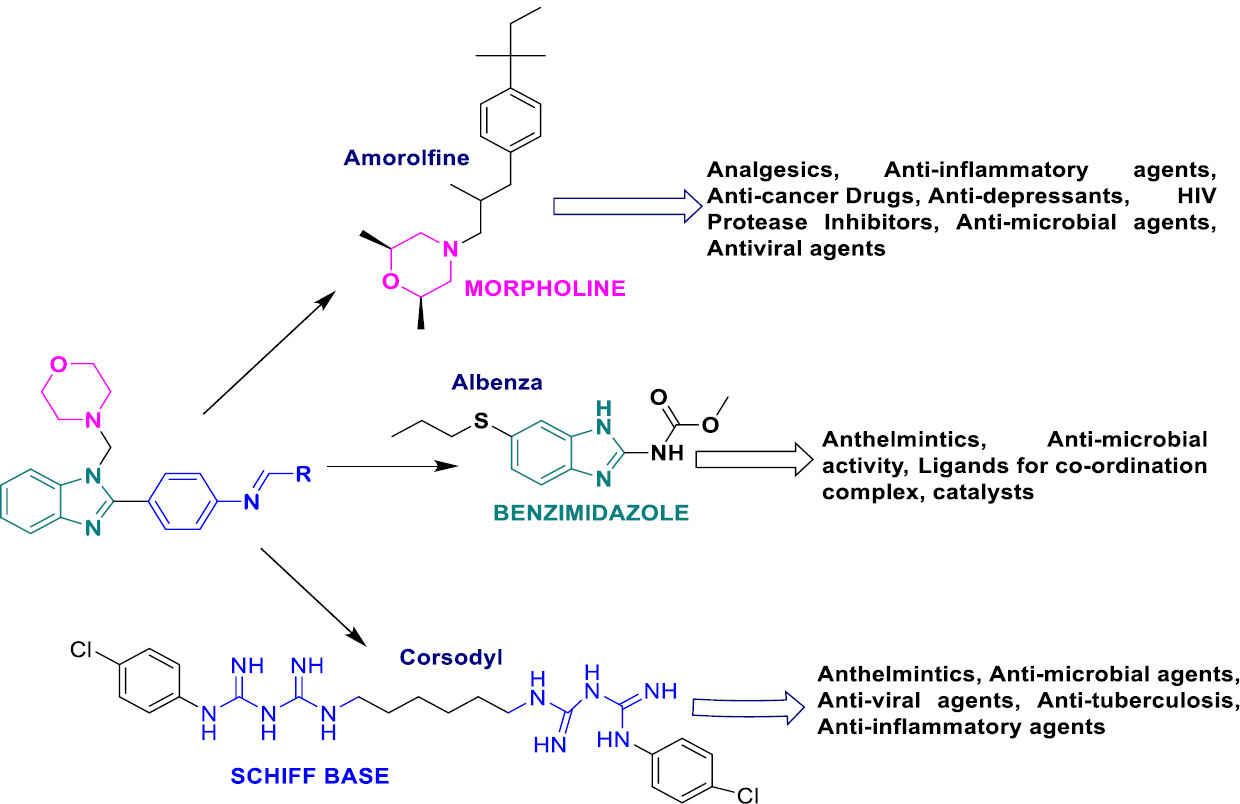
Benzimidazoles have received a lot of attention because of their diverse biological activities and structural variety [11-13]. The synthesis of multiple functional groups, along with benzimidazole, allows for the customization of characteristics to specific targets, and the addition of Schiff base and morpholine core enhances their bioactivity. Benzimidazole scaffolds are renowned antibacterial, antifungal, antiviral, and anticancer agents. Their targeted actions frequently disrupt the principle of cellular progress during infection and make them vulnerable to eradication [14, 15]. By sculpting this benzimidazole motif in its favorable positions and combining it with other essential cores, we can further enhance its potency and create promising candidates for the development of novel antimicrobial medicines.
In this context, Ya Yan et al. synthesized novel benzimidazole moieties on fusion with 4-oxyquinazoline for enhancement of their bioactivity, which offers potential applications in combating microbial infections. Their findings underscore the importance of hybrid molecular scaffolds in developing next-generation antimicrobials and anti-cancer agents. Being antimicrobial, benzimidazoles disrupt microbial cell wall synthesis and act as anticancer agents that target specific pathways for the inhibition of cellular proliferation and apoptosis. Hence, they suggested that the adaptability of benzimidazole in chemical modification enables it to address a wide range of activities, such as antimicrobial, antiviral, anti-inflammatory, and antiparasitic effects. Consequently, it is a critical scaffold for drug development in a variety of therapeutic areas. The benzimidazole scaffold rapidly interacts with biological substrates through non-covalent forces, such as hydrogen bonds, hydrophobic effects, π-π stacking, and van der Waals interactions. Hence, some meaningful fusion over the benzimidazole nucleus could provide enhancement in single and multifunctional pharmacological activities [16].
The literature found that Schiff bases fused with benzimidazole cores have antiparasitic and antimicrobial activities, as well as cytotoxic effects on some cancer cells. Generally, schiff bases exhibit their biological effects with anti-bacterial and anti-inflammatory properties [11, 14, 17]. This excellent result encouraged researchers to further develop lead molecules as antibacterial agents [15]. However, researchers suggested that a group of arylidenimidazolones with morpholine substitutions at positions 2 and 5 could be useful for adding antimicrobials to antibiotic therapy [18]. Due to their ease of synthetic accessibility and configurational changes, they initiate vital modules in the drug discovery process. Furthermore, the morpholine core has a lot of antibacterial power by breaking down membrane integrity, slowing down pathogens' nucleic acid production, and blocking enzymes [19-21]. In this study, adding Schiff base and morpholine motifs to the benzimidazole scaffolds enhanced the biological activity in novel ways [22].
Computational approaches to drug discovery have altered the overall scenario by narrowing down our research hypotheses. This computational intelligence speeds up the search for lead compounds with high binding affinities and better pharmacokinetic characteristics. The three possible targets were preferably chosen for the prediction of biomolecules on bacterial, fungal, and resistant microbial pathogens for the determination of antimicrobial effects. The first target is enoyl-acyl carrier protein (ACP) reductase (PDB.ID: 1C14), a vital enzyme involved in the biosynthesis of bacterial fatty acids. The conversion of trans-2,3-enoyl-ACP to acyl-ACP is a peculiar process that aims to elongate fatty acid synthesis [11]. Hence, inhibition of this enzyme is useful for investigating potential antibiotic candidates, particularly against gram-negative bacteria, such as Escherichia coli (E. coli) [14]. The second target is glucosamine-6-phosphate synthase (PDB.ID: 1JXA), which is an important enzyme that converts fructose-6-phosphate and glutamine into glucosamine-6-phosphate. It is an important step in the process of peptidoglycan biosynthesis [15]. Consequently, inhibition of this enzyme affects cell wall production, and it is a potential target for both antibacterial and antifungal agents [18].
Certainly, for the development of antimicrobial agents, consideration of microbial resistance is essential in drug discovery. In this regard, the third target selected is D-Alanyl-D-Alanine Dipeptidase (PDB.ID: 1R44) because antibiotic-resistant bacteria tend to change the composition of cell wall materials, including the peptidoglycan layer [11]. Beta-lactam antibiotics disrupt cell wall production by targeting D-Alanyl-D-Alanine dipeptidase. Mutations of this target enzyme may impair the efficiency of these medicines, which leads to antibiotic-resistant bacteria [14]. The goal is to create compounds that block this enzyme, preventing peptidoglycan production and making bacteria more susceptible to antibiotics or the host's immune system. Hence, the D-Alanyl-D-Alanine dipeptidase inhibitors in our titled compounds could prevent resistance mechanisms and improve the efficacy of the molecules.
The fusion of benzimidazole, Schiff base, and morpholine motifs can synergistically work together to produce better bioactivity profiles (Fig. 1). The smart combination of computer predictions and synthetic techniques makes it possible to create a focused library of compounds that have promising biological potency in addition to fighting pathogens that are resistant to drugs. The research also aims to uncover the structure-activity relationship of these novel molecules, which could offer a lot of information on these notable molecular features that influence their antibacterial potency. This study provides a framework for future advances in medicinal chemistry and the discovery of antimicrobial agents.
2. MATERIALS AND METHODS
2.1. Protein Preparation
The x-ray crystal structures of targeted enzymes, such as Enoyl ACP reductase of E.coli (PDB.ID: 1C14), Glucosamine-6- phosphate synthase (PDB.ID: 1JXA), and D-alanyl-D-alanine-dipeptidase(PDB.ID: 1R44) were collected from protein data bank of research Collaboratory for structural bioinformatics portal (http://www.rcsb.pdb.org/). The selected proteins were pre-processed and purified by deletion of water, unwanted heteroatoms, and repair of missing residues by the Molegro molecular viewer (MMV 2012-2.5.0 version) [23, 24].
2.2. Ligand Preparation
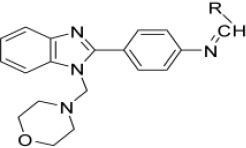
The PDB files of synthesized N-substituted-benzylidene-4-(1-(morpholinomethyl)-1H-benzimidazol-2-yl)benzamine (3a-3o) (Table 1) test compounds were prepared from chem 3D-pro 12.1 software tool [25]. Those compounds were pre-processed and optimised by minimizing their energy and adding Gasteiger charges and polar hydrogens, and torsions were set from Autodock 4.2. software (http://autodock.scripps.edu/) [26].
Designed benzimidazole derivatives.
| General Structure for Synthesized Compounds | ||
|---|---|---|
 |
||
| Code | R | IUPAC Name of the Compounds |
| 3a | -
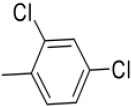 |
N-(2,4-Dichlorobenzylidene)-4-(1-(morpholinomethyl)-1H-benzoimidazol-2-yl) benzenamine |
| 3b | -
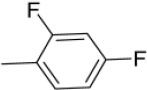 |
N-(2,4-Difluorobenzylidene)-4-(1-(morpholinomethyl)-1H-benzimidazol-2-yl)benzenamine |
| 3c | -
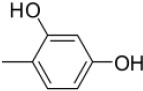 |
4-((4-(1-(Morpholinomethyl)-1H-benzimidazol-2-yl)phenylimino)methyl)benzene-1,3-diol |
| 3d | -
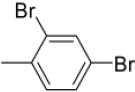 |
N-(2,4-Dibromobenzylidene)-4-(1-(morpholinomethyl)-1H-benzimidazol-2-yl)benzenamine |
| 3e | -
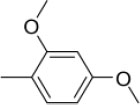 |
N-(2,4-Dimethoxybenzylidene)-4-(1-(morpholinomethyl)-1H-benzimidazol-2-yl) benzenamine |
| 3f | -
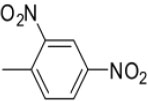 |
N-(2,4-Dinitrobenzylidene)-4-(1-(morpholinomethyl)-1H-benzimidazol-2-yl)benzenamine |
| 3g | -
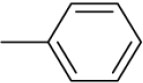 |
N-Benzylidene-4-(1-(morpholinomethyl)-1H-benzimidazol-2-yl)benzenamine |
| 3h | -
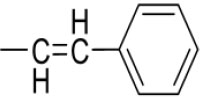 |
4-(1-(Morpholinomethyl)-1H-benzimidazol-2-yl)-N-(3-phenylallylidene)benzenamine |
| 3i | -
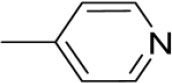 |
4-(1-(Morpholinomethyl)-1H-benzimidazol-2-yl)-N-(pyridin-4-ylmethylene)benzenamine |
| 3j | -
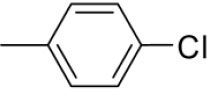 |
N-(4-Chlorobenzylidene)-4-(1-(morpholinomethyl)-1H-benzoimidazol-2-yl)benzenamine |
| 3k | -
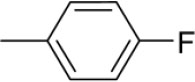 |
N-(4-Fluorobenzylidene)-4-(1-(morpholinomethyl)-1H-benzimidazol-2-yl)benzenamine |
| 3l | -
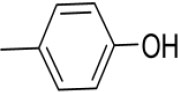 |
4-((4-(1-(Morpholinomethyl)-1H-benzimidazol-2-yl)phenylimino)methyl)phenol |
| 3m | -
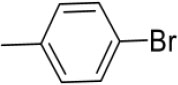 |
N-(4-Bromobenzylidene)-4-(1-(morpholinomethyl)-1H-benzimidazol-2-yl)benzenamine |
| 3n | -
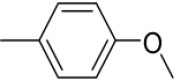 |
N-(4-Methoxybenzylidene)-4-(1-(morpholinomethyl)-1H-benzimidazol-2-yl)benzenamine |
| 3o | -
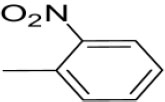 |
N-(2-nitrobenzylidene)-4-(1-(morpholinomethyl)-1H-benzimidazol-2-yl)benzenamine |
2.3. Docking Protocol
The refined proteins and ligands (energy minimized) were assessed for molecular simulation to predict their binding affinity and key residual sites over the enzyme. The grid map was set with 90 points, and the Lamarckian genetic algorithm was accomplished with 25,000,000 energy evaluations [27]. For each run, 5,000 generations were done, and 150 docking runs were achieved [28, 29]. The binding poses for docked compounds were visualized by Biovia, Discovery studio visualiser programme [30].
2.4. Lead Optimisation
The effectiveness of synthesized moiety on pharmacokinetic profile (ADME), physicochemical properties, and toxicity profile [31-33] was analysed by using Qikprop in Schrodinger LLC’s Programme version 4.4 [34].
2.5. Density Functional Theory
The GaussView molecular visualised programme and the Gaussian 03W package were used to perform computational computations on the synthesised compounds. Density functional theory (DFT) was used to optimize the ground state of molecular structures of synthesised compounds by employing B3LYP functional with a basic set of 6-311G (d,p). The optimised structures of synthesized compounds were used to calculate the frontier molecular orbital energies (lowest unoccupied molecular orbital [ELUMO], highest occupied molecular orbital [EHOMO], and their energy gap [32, 35]) of all molecules. GaussView molecular visualised programme was used to visualise the acquired molecular orbital energy diagrams of the synthesised compounds [36, 37].
2.6. Synthetic Procedures
The titled compounds were synthesized in three steps. The first step was the general method of benzimidazole synthesis (Compound 1) [38], followed by a mannich base reaction for the fusion of morpholine with benzimidazole motif (Compound 2) [39, 40]. These experimental procedures and spectroscopic and analytical data are provided in supplementary details (S1-S3). Finally, compound (2) was initiated for Schiff base reaction with substituted aromatic aldehydes to form titled compound (3a-o).
2.7. Synthesis of N-substitutedbenzylidene-4-(1-(morpholinomethyl)-1H-benzimidazol-2-yl) benzamine (3a-o) [41]
The equimolar of 4-(1-(morpholinomethyl)-1H-benzimidazol-2-yl)benzenamine (3.08 gm) and different aromatic aldehyde in ethanol (50 mL) was taken in a round-bottomed flask. To this, glacial acetic acid (1 mL) was added and refluxed for 10 h and kept aside for one day. Then, the solution was poured into ice-cold water, stirred well, and the separated product was filtered. The dried product was recrystallized using ethanol. The characteristic details of synthesized compounds for 3a-3o are mentioned in Supplementary S3, S4, and the scheme of synthesis in Fig. (2).
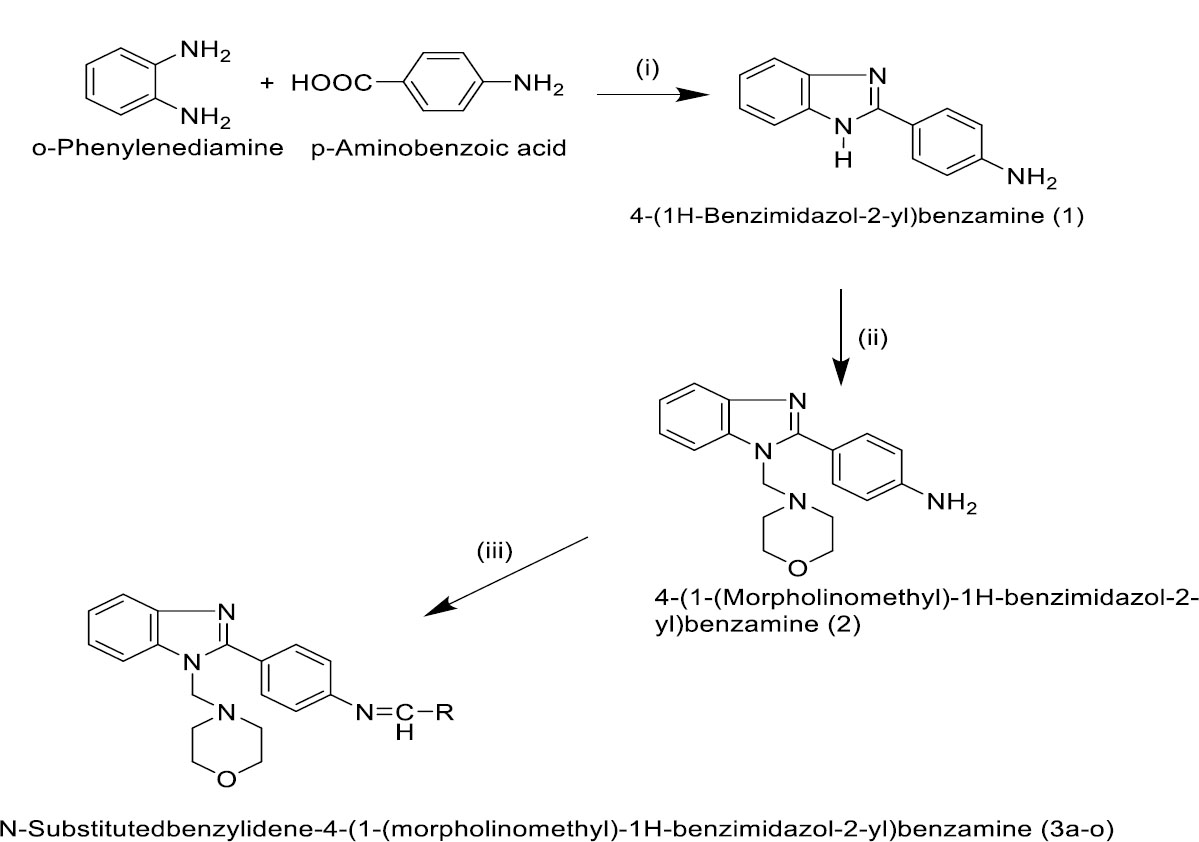
Synthesis of benzimidazole derivatives (3a-3o).Reagents and conditions: (i) Concentrated HCl, Stir, and reflux in the water bath for 2 h (ii) morpholine and formaldehyde, stir 2h, reflux on the water bath for 4 h; (iii) RCHO, CH3COOH, pH 4-5, reflux for 10 h, 3a-o.
2.8. Biological Evaluation
2.8.1. Antimicrobial Activity
The synthesized compounds (3a-3o) with a concentration of 50 µg/mL were assessed for antimicrobial activity by agar diffusion method against three gram-positive and gram-negative bacterial strains and on two strains of fungal pathogens [42]. Mueller Hinton agar plates and sabouraud dextrose agar medium with a thickness of 5-6 mm were made aseptically for bacteria and fungi pathogens, respectively. The prepared plates were acceptable for solidification and dried before inoculation at 37°C. The sterile swabs used for inoculation were dipped over the medium plates and streaked three times on 360° rotation for each application. After streaking, medium plates closed with lids were subjected to drying at room temperature. With the aid of sterilised cork borers, wells were made in which synthesized compounds and standard drugs were added by micropipettes, followed by refrigeration (1 h) to initiate uniform diffusion of added compounds. Finally, those mediums were incubated at 37 °C for 18 to 24 h and zone of inhibition for tested compounds was measured and compared with a concentration of 5 µg/mL for Gatifloxacin (anti-bacterial) and Clotrimazole (anti-fungal), respectively as reference drug. By considering the zone of inhibition of standard as 100% and by using the following formula, the percentage inhibition (PI) of synthesized compounds was calculated.
PI of synthesized compounds = (Zone of inhibition of synthesized compound / Zone of inhibition of respective Standard) x 100
2.8.2. Minimum Inhibitory Concentration
The synthesized compounds were subjected to the determination of minimum inhibitory concentration (MIC) by a two-fold serial dilution method [43]. The synthesized derivatives (3a-3o) were exposed for the preparation of primary stock solution (1000 µg/mL) by using DMSO solvent. The nutrient broth (Bacteria) and sabouraud dextrose broth (Fungi) were utilized for the preparation of seeded broth from 24 h old bacterial culture and 1-7 days old culture on nutrient agar and sabouraud agar, respectively. The plating technique was used to achieve 113-115 colony-forming units (cfu/mL) of seeded broth. The final inoculum size for the antibacterial assay was 114 cfu/mL, and for the antifungal assay, it was 111 cfu/mL, and pH was maintained appropriately for bacteria at 7.4 ± 0.2, and for fungi, it was 5.6.
From the primary stock solution, the first dilution was done by the addition of 0.5 mL on 1.5 mL of seeded broth and considered as a secondary stock solution [44]. By using this secondary stock, a further 1 mL was pipetted out and suspended on 1 mL of seeded broth. The same procedure was followed for the preparation of assay tubes. Up to eight dilutions were completed. Further, these assay tubes were incubated at 37±1°C (bacteria) and 28±1°C (fungi), and control was considered by seeded broth tubes. The reference drugs were prepared accordingly for both antibacterial (gatifloxacin) and antifungal (Clotrimazole) activities [45, 46].
2.9. Statistical Analysis
The results were stated as mean ± SD on three parallel measurements. ONE-WAY ANOVA followed by Dunnett’s test was utilised for differences between control tubes and test tubes by using Graphpad-Instat software [47].
3. RESULTS AND DISCUSSION
3.1. Chemistry
The starting reactants of chemical compounds, reagents, and solvents were procured from Sigma Aldrich, Himedia Ltd, and Merck Ltd, and those were HPLC-graded components [48]. The evaluation of melting point for synthesized compounds was performed by digital melting point apparatus by open capillary approach. The chromatogram for synthesized compounds was performed by Silica gel G TLC plates (Merck Ltd) and it was detected by an iodine chamber. Further, various spectroscopic techniques were utilized for the characterization of synthesized compounds, such as λ max (Schimadzu Ultraviolet spectrometer), IR spectrum (Schimadzu FT-IR spectrophotometer) using KBr pellets, elemental analysis (CHNO Analyzer), mass spectrum (HRMS mass spectrometer), and 1HNMR spectrum (FT-NMR 500 MHz spectrometer). These analyses were performed by the Indian Institute of Technology, Chennai, and VIT University, Vellore.
Novel N-substitutedbenzylidene-4-(1-(morpholinomethyl)-1H-benzimidazol-2-yl)benzamines (3a-3o) were synthesized using multi-step synthesis according to the protocol specified in the synthetic scheme (Fig. 2). Initially, o-phenylenediamine and p-aminobenzoic acids were cyclized in the presence of concentrated hydrochloric acid to produce 4-(1H-benzimidazol-2-yl)benzamine (1). In the next step, 4-(1H-benzimidazol-2-yl)benzamine (1) was treated with morpholine and formaldehyde to produce 4-(1-(morpholinomethyl)-1H-benzimidazol-2-yl)benzamine (2) by Mannich base reaction.
Finally, title compounds were prepared by treating 4-(1-(morpholinomethyl)-1H-benzimidazol-2-yl)benzamine (2) with different aromatic aldehydes in the presence of acetic acid by Schiff base reaction. TLC was employed for the reaction optimization, completion, and purity of the prepared title and intermediate derivatives. The mechanism of these three steps is illustrated in Fig. (3A-3C).
3.1.1. First Step (Ring closure reaction)
Initially, the carboxylic group of p-amino benzoic acid was protonated using hydrochloric acid to produce activated p-amino benzoic acid. Later, this activated p-amino benzoic acid reacted with the nitrogen of one of the amino groups in o-phenylene diamine and produced a quaternary nitrogen derivative. Subsequently, quaternary nitrogen underwent deprotonation and produced secondary amino compounds. Then, protonation occurred at one of the hydroxyl groups, followed by dehydration, producing intermediate carbocation analogs. In the next step, deprotonation of the hydroxyl group produced corresponding ketone analogs. Later, the amide group (keto form) underwent keto-enol tautomerism and produced the enol form of the amide group. Finally, intramolecular cyclization produced a five-membered benzimidazole ring system with a loss of water. The detailed reaction mechanism is mentioned in Fig. (3A).
3.1.2. Second Step (Mannich base reaction)
In the second step, the keto group of formaldehyde was initially protonated and produced activated carbocation. Later, this activated carbocation reacted with the nitrogen of one of the amino groups present in benzimidazole and produced respective quaternary nitrogen derivatives. Subsequently, quaternary nitrogen underwent deprotonation and produced tertiary amino compounds. Later, protonation took place in the hydroxyl group, followed by dehydration, producing intermediate carbocation analogs. In the next step, this carbocation was attacked by a lone pair of electrons present in the nitrogen of the morpholine ring and produced a quaternary nitrogen derivative. Finally, the quaternary nitrogen derivative was deprotonated and produced Mannich base derivatives of benzimidazole [49]. The detailed reaction mechanism is mentioned in Fig. (3B).
3.1.3. Third Step (Schiff base reaction)
Initially, the keto group of aromatic aldehydes was protonated and produced activated carbocation. Later, this activated carbocation reacted with the nitrogen of the amino group present in 4-(1-(morpholinomethyl)-1H-benzimidazol-2-yl)benzamine (2) and produced a quaternary nitrogen derivative. Then, protonation occurred in the hydroxyl group, followed by dehydration-producing intermediate carbocation analogs. In the next step, this carbocation was attacked by a lone pair of electrons present in the nitrogen of the amino group and produced respective quaternary nitrogen derivatives. At last, quaternary nitrogen underwent deprotonation and produced Schiff base derivatives of benzimidazole [50]. The detailed reaction mechanism is mentioned in Fig. (3C).
The percentage yield of all synthesized compounds (3a-3o) ranged from moderate to good (70-81%w/w). The melting point of the synthesized compounds was noted in the range of 141-201 °C, and reaction progress was recorded by using thin-layer chromatography by observing a single spot, which was detected by the iodine chamber. The elemental analysis of synthesized compounds revealed that the calculated value resembled the accuracy of the molecular formula for those compounds. The mass spectrum of the synthesized compounds (3a-3o) indicated the molecular ion peak as (M+) m/z: 464.2110 (M+), 432.0271 (M+), 428.1214 (M+), 552.1017 (M+), 456.5338 (M+), 486.1065 (M+), 396.1357 (M+), 422.3422 (M+), 397.2314 (M+), 430.1464 (M+), 414.1426 (M+), 412.2411 (M+), 474.1325 (M+), 426.5338 (M+), and 441.0431 (M+), respectively and this confirmed the molecular weight of synthesized compounds.
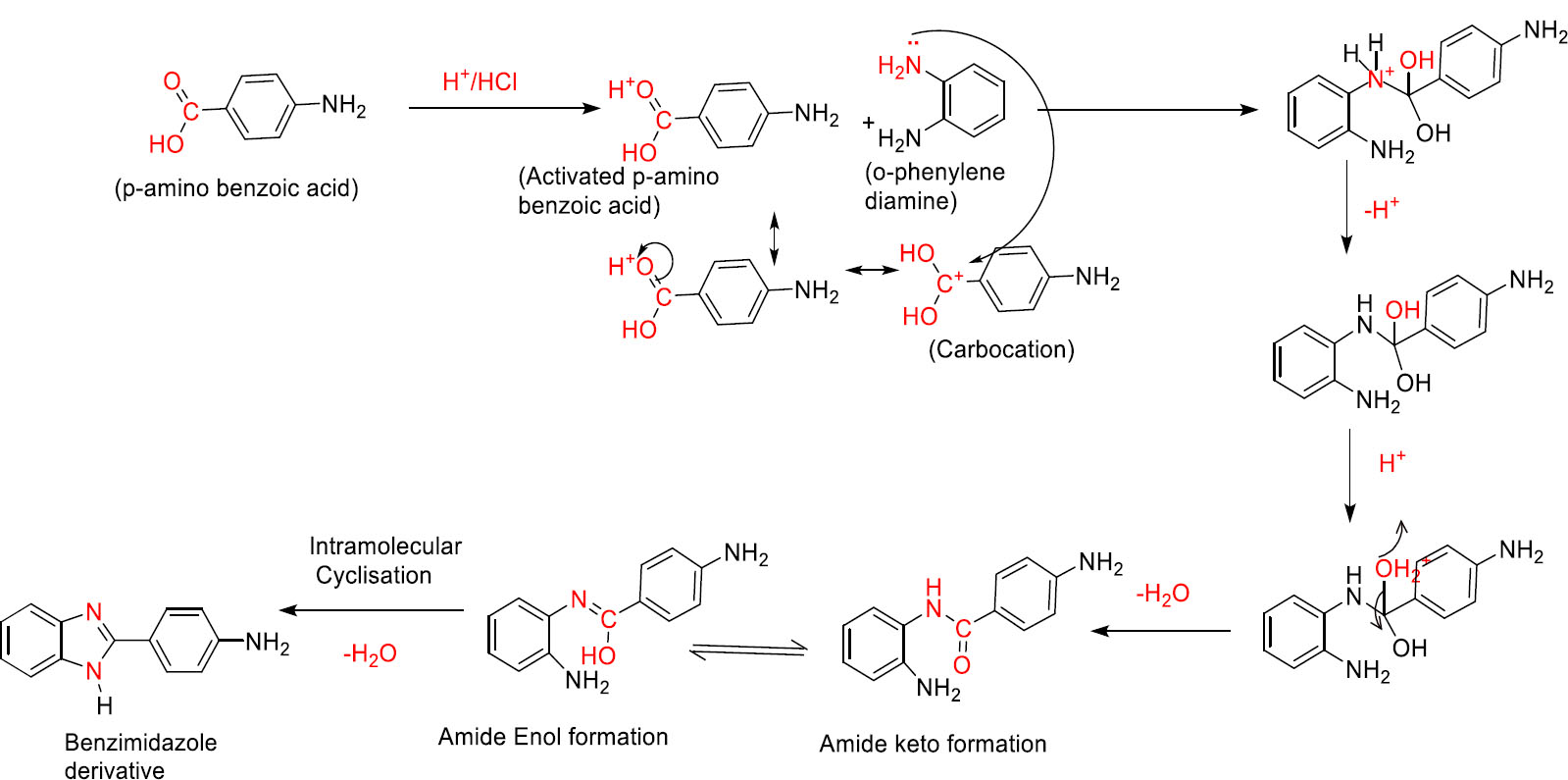
Mechanism for synthesis of benzimidazole with para-amino benzaldehyde.
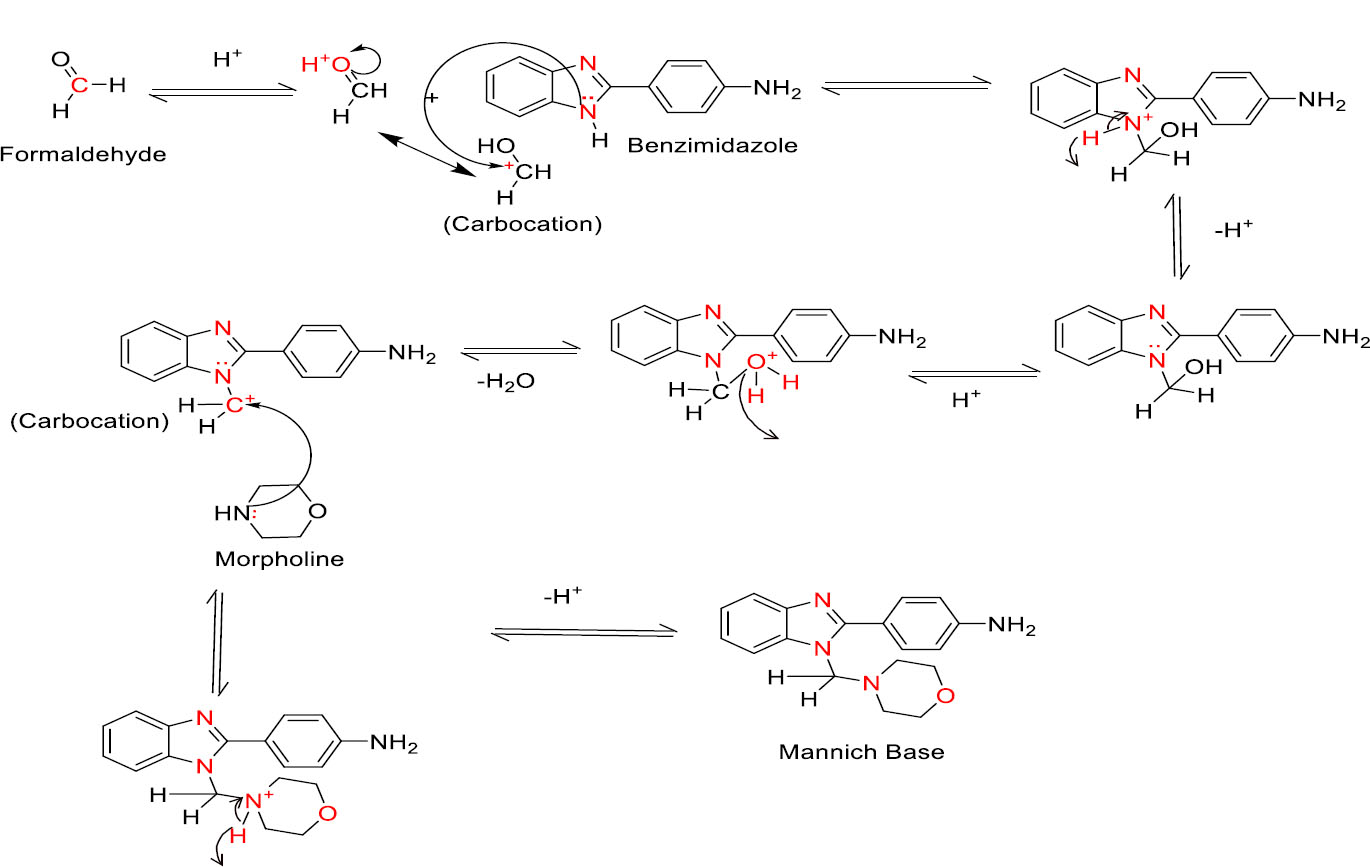
Mechanism of fusion of morpholine with benzimidazole.
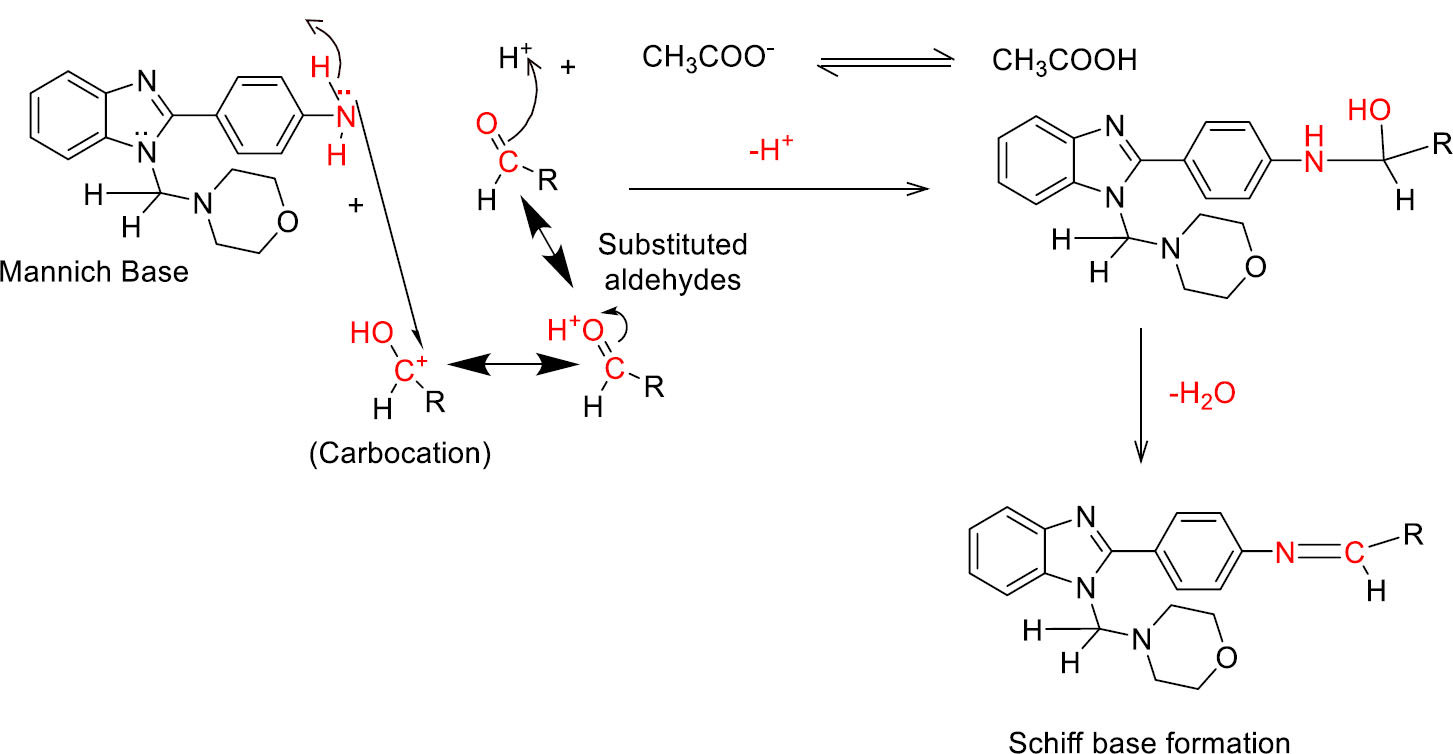
Mechanism of Schiff base formation.
The chemical structure of the prepared intermediates and title compounds were confirmed by various spectroscopic techniques, such as IR, 1H-NMR, 13C-NMR mass spectra, and microanalysis. Spectral studies of all compounds confirmed the assigned structures. Characteristic absorption peaks present in IR spectra represented the presence of some specific groups in chemical structure. In the IR spectrum of 4-(1H-benzimidazol-2-yl)benzamine (1), the appearance of an absorption peak at 3374 cm-1 corresponded to NH stretching, and the disappearance of the absorption peak around 3500 cm-1 corresponded to carboxylic acid confirmed its formation. It was further supported by the appearance of a singlet peak in the NMR spectrum at δ 5.41 ppm, which corresponded to two protons of NH2, and the appearance of a singlet peak in the NMR spectrum at δ 4.76 ppm, which corresponded to one proton of NH of benzimidazole. In the IR spectrum of 4-(1-(morpholinomethyl)-1H-benzimidazol-2-yl)benzamine (2), the appearance of an absorption peak at 2938 cm-1 corresponded to CH2-H stretching and the appearance of an absorption peak around 1059 cm-1 corresponded to C-O-C, which confirmed its formation. It was further supported by the disappearance of a singlet peak in the NMR spectrum at δ 4.76 ppm, which corresponded to one proton of NH of benzimidazole. In addition, the appearance of a triplet peak in the NMR spectrum at 2.71 ppm corresponded to four protons of CH2 of morpholine and at 3.58 ppm, which corresponded to four protons of CH2 of morpholine. The appearance of a singlet peak in the NMR spectrum at 5.41 ppm corresponded to two protons of CH2 linkage and also confirmed the formation of compound (2). In the IR spectrum of N-substitutedbenzylidene-4-(1-(morpholinomethyl)-1H-benzimidazol-2-yl)benzamine (3a-o), the disappearance of the absorption peak at 3300 cm-1 corresponded to NH stretching and disappearance of singlet peak in NMR spectrum at δ 5.41 ppm corresponded to two protons of NH2, which confirmed its formation. It was further supported by the appearance of a singlet peak in the NMR spectrum around 8.50 ppm, corresponding to one proton of -N=CH-.
All the titled compounds (3a-3o) showed their characteristic peaks in the region of 1033-1083 cm-1, 1605-1645 cm-1, 1642-1679 cm-1, 2874-2948 cm-1, and 3005-3039 cm-1, which corresponded to C-O-C stretching, C=C in aromatic rings, C=N stretching, CH2-CH stretching, and aromatic C-H stretching, respectively. The 1H and 13C-NMR of synthesized compounds confirmed the presence of aromatic rings by the formation of multiplets in the range of 6.72-8.67 δ ppm and 150–114 δ ppm, respectively, due to the presence of nearby functional groups or substituents. The presence of the imine group, confirmed by the formation of singlet at 8.31-8.86 δ ppm (1H-NMR) and 169–155 δ ppm (13C-NMR), indicated the presence of carbons in different environments, including carbonyl, imine group (N=CH) and few aromatic carbons. In both spectrums, methylene groups associated with morpholine ring were detected by triplets at two ranges for four hydrogens, each in the range of 2.02-2.97 δ ppm and 3.31-3.95 δ ppm (1H-NMR). In the case of 13C-NMR, two distinct ranges were observed for each carbon atom, between 72–59 δ ppm and 57–40 δ ppm, respectively.
The presence of methoxy groups was confirmed by singlet at 3.83-4.12 δ ppm for compounds 3e and 3n (1H-NMR) and for 13C-NMR 58-52 δ ppm, respectively. In addition, the presence of -CH2- linkage was detected by singlet at 4.63-5.27 δ ppm (1H-NMR) and 73-60 δ ppm (13C-NMR). The overview of various spectral analyses clearly confirms the presence of aromatic rings, the imine group, -CH2- linkage, and morpholine rings in all synthesized compounds.
In the NMR spectrum, the appearance of other characteristic peaks also supported the proposed structure of synthesized derivatives. Further mass spectrum confirmed their purity and molecular weight. In addition, micro-analysis reports were also within the limit, further supporting the proposed chemical structure of the synthesized analogs.
3.2. Molecular Docking Studies
The path of drug development as antimicrobial agents could be crucial from a pathophysiological view because it is responsible for developing antimicrobial properties. Hence, the selection of targets should be essential for the development of antimicrobials as a bactericidal and fungicidal agents. Henceforth, synthesized compounds were predicted for three different targets for the inhibition of both bacterial and fungi pathogens [51, 52].
The molecular docking simulations were performed on synthesized N-substitutedbenzylidene-4-(1-(morpholinomethyl)-1H-benzimidazol-2-yl) benzamine (3a-3o) by multitargeted proteins for antimicrobial activities. They were Enoyl Acyl Carrier Protein reductase of E.coli (PDB.ID: 1C14), Glucosamine-6- phosphate synthase (PDB.ID: 1JXA), and D-Alanine: D-Alanine Ligase with ATP from Thermus thermophius (PDB.ID: 2ZDQ) [11, 14, 15].
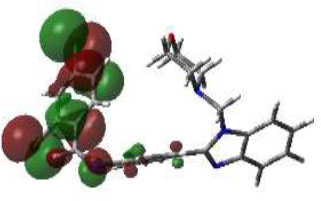
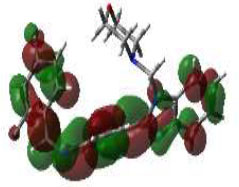
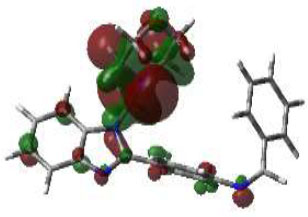
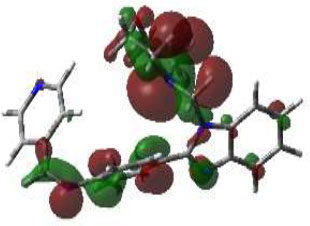
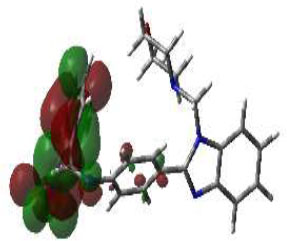
From this perspective, the first target was the Enoyl Acyl Carrier Protein reductase of E. coli, and it is quite responsible for the biosynthesis of fatty acids for the development of the cellular wall of the microbes [53]. Hence, the cellular wall of microbes is helpful for their survival by their active participation in the progression of infection. The important components of fatty acids present on the cellular wall are lipopolysaccharides and phospholipids. The biosynthetic pathway begins with acetylcholine and continues with several stages with the involvement of various enzymes. But Enoyl ACP reductase is involved in the termination step, and it is helpful for the conversion of trans-2-enoyl ACP to acyl ACP. This component is essential for the building up of microbial cell walls as a phospholipid biosynthetic component [18]. The docking score of synthesized compounds on Enoyl ACP reductase [1C14] was recorded and is presented in Table 2. All the synthesized compounds produced affordable results by producing low binding energy, which denoted lower bonding energy between the ligand and targeted enzyme illustrated [19]. The docking scores were noted from -5.27 to -8.76 Kcal/mole and those produced excellent hydrogen bonding and steric interaction with their amino acid residues. Among the synthesized compounds, compound (3e), N-(2,4-Dimethoxy-benzylidene)-4-(1-(morpholino-methyl)-1H-benzimidazol-2-yl)benzene amine, afforded an excellent binding score of -8.76 kcal/mol, compared to the reference drug, which had a score of -7.13 kcal/mol. Its interactions are shown in Fig. (4A). The binding pocket of compound 3e on enoyl acyl carrier protein reductase demonstrated conventional hydrogen bonding interactions (Asn A 155 & Tyr A 104) and steric interactions, including pi-anion (Asp A 103 & Ala B 146) and pi-alkyl (Pro A 154, Pro B 1179, Ala B 152 & 1176) with amino acid residues.
The second target was glucosamine-6-phosphate synthase (1JXA), a complex enzyme that catalyzes the conversion of L-glutamine into fructose-6-phosphate via ammonia transfer. This is followed by isomerization, leading to the formation of fructosamine-6-phosphate, which is then converted into glucosamine-6-phosphate. This reaction is essential for bacterial and fungal pathogens, aiding in the assembly of cell walls and the formation of macromolecules, such as chitin and mannoproteins (in fungi) and peptidoglycan (in bacteria) [54]. Due to this reason, it represents a potential target for antifungal agents, and even short-term inhibition of glucosamine-6-phosphate synthetase can lead to changes in morphology, agglutination, and lysis of pathogens, which are lethal to fungal organisms [20, 21].
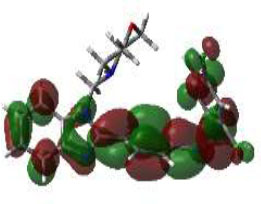
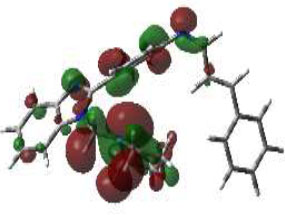
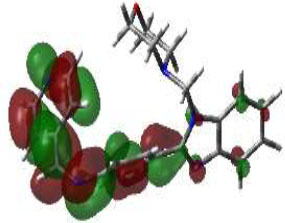
Hence, in the screening of anti-microbial activity on synthesized benzimidazole derivatives, this target was selected and evaluated for docking studies. The synthesized compounds afforded excellent binding scores ranging from -4.85 to -9.91 kcal/mol (Table 2), with compound 3b, N-(2,4-Difluoro-benzylidene)-4-(1-(morpholino-methyl)-1H-benzimidazol-2-yl)benzenamine, producing the highest binding score of -9.91 kcal/mol compared to the reference drug. The active site of compound 3b showed conventional hydrogen bond interactions with Val A 570 and Ser A 316, pi-anion interactions with Asp A 432, amide-pi-stacked interactions with Gly A 473, pi-alkyl interactions with Val A 510, and halogen (fluorine) interactions with Asp A 474, as represented in Fig. (4B).
The third targeted enzyme was D-alanine ligase responsible for chemical reaction with ATP to form D-alanyl D-alanine, ADP, and Phosphate molecules [55]. This enzyme is essential for inhibition of bacterial cell walls and particularly for peptidoglycan biosynthesis. Most of the antibiotics from penicillin onwards could be a universal target for bacteria cell walls by peptidoglycan biosynthesis inhibition.
The targeted receptor comes under the ATP family and leads to the phosphorylation of D-alanine bound substrate, forming an intermediate named phosphoryl carboxylate, which is prone to nucleophilic attack by its carbon-nitrogen terminus (D-amino acid), which is considered a catalytic domain [56]. As a result, dipeptides are formed, which could be merged on peptidoglycan as a tripeptide chain with the aid of the next enzyme in the cytoplasm of the biosynthetic pathway of peptidoglycan [57]. The anti-tubercular drug (D-cycloserine) and antibiotic (Vancomycin) are familiar drugs used to inhibit bacterial cell wall synthesis by inhibition of D-ala-D-ala ligase.
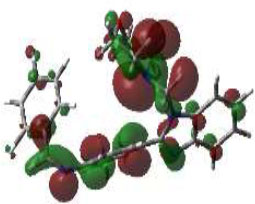
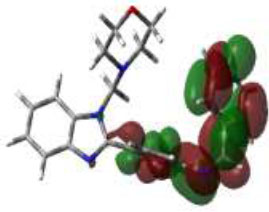
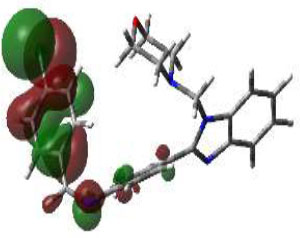
Due to the hydrogen bond interaction of vancomycin on terminal D-ala-D-ala ligase, it inhibits the reaction of transpeptidation in peptidoglycan polymers. Apart from that, vancomycin resistance has emerged; in these strains, D-ala-D-ala lactate was replaced with a terminal chain of D-Ala-D-Ala on peptidoglycan polymers. By which vancomycin binding on that targeted protein was prevented., which leads to the formation of resistant [58]. Due to the dual role of this protein, the synthesized compounds were screened for molecular docking study. Of the fifteen compounds, 3f afforded high potential binding of -7.75 kcal/mole compared to cycloserine, with a score of -6.97 kcal/mol, as shown in Table 2. Hydrophilic and hydrophobic interactions of compound 3f were observed with amino acid residues through hydrogen bonding with Pro B 67 and 68, amide-pi-stacked interactions with Trp B 71, and pi-alkyl interactions with Leu B 98, as shown in Fig. (4C).
| Compound Code |
Docking Score (ΔG Kcal/Mol) |
||
|---|---|---|---|
| [PDB:ID: 1C14] | [PDB:ID: 1JXA] | [PDB:ID: 2ZDQ] | |
| 3a | -7.48 | -7.62 | -5.48 |
| 3b | -8.45 | -9.91 | -6.36 |
| 3c | -7.76 | -7.73 | -5.43 |
| 3d | -8.31 | -8.57 | -7.42 |
| 3e | -8.76 | -5.72 | -5.61 |
| 3f | -6.66 | -9.01 | -7.55 |
| 3g | -7.77 | -6.29 | -5.05 |
| 3h | -8.32 | -8.77 | -6.47 |
| 3i | -5.66 | -4.93 | -5.38 |
| 3j | -7.03 | -8.83 | -6.08 |
| 3k | -7.8 | -6.97 | -5.5 |
| 3l | -7.76 | -5.61 | -6.56 |
| 3m | -7.64 | -7.02 | -7.7 |
| 3n | -8.44 | -7.04 | -7.53 |
| 3o | -5.27 | -4.85 | -6.97 |
| Standard | -7.13* | -7.13* | -6.97 |
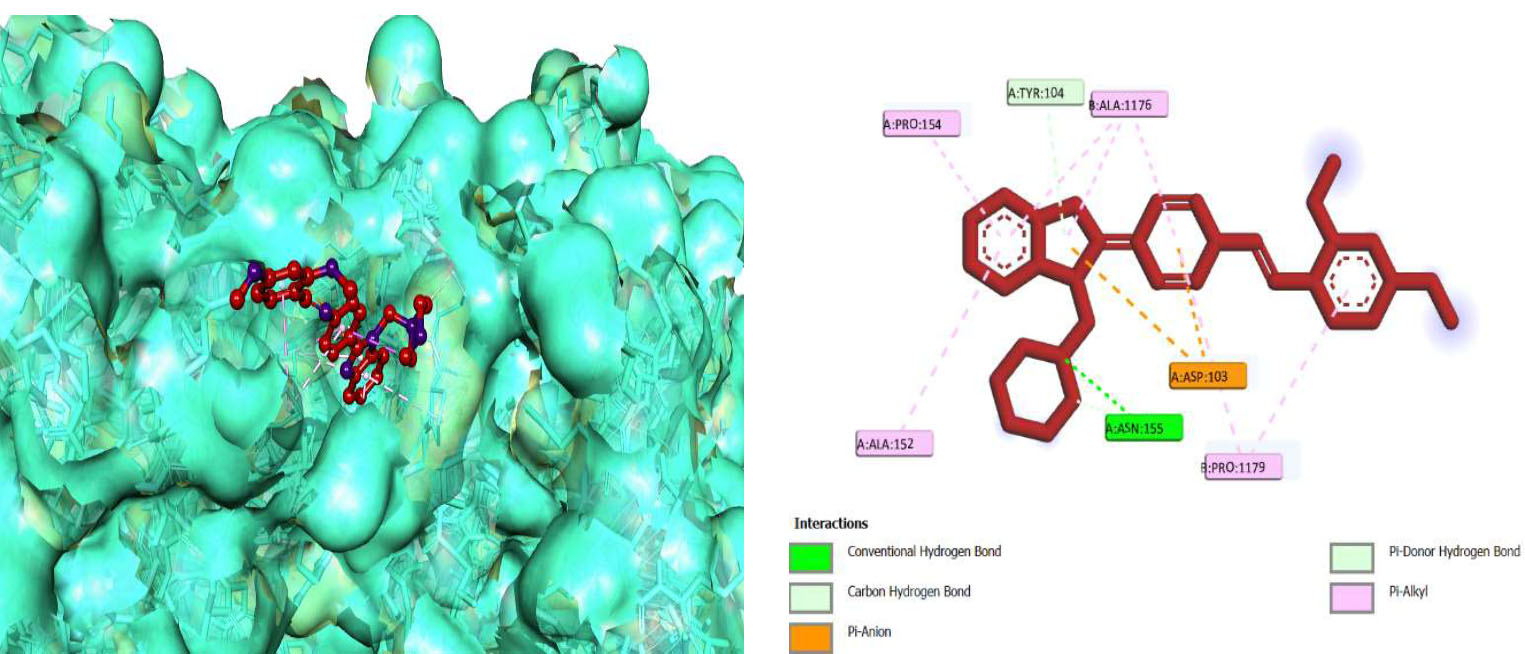
The 3D docked pose and 2D dimension of tested compound 3e on Enoyl ACP reductase of E. coli (PDB ID: 1C14).
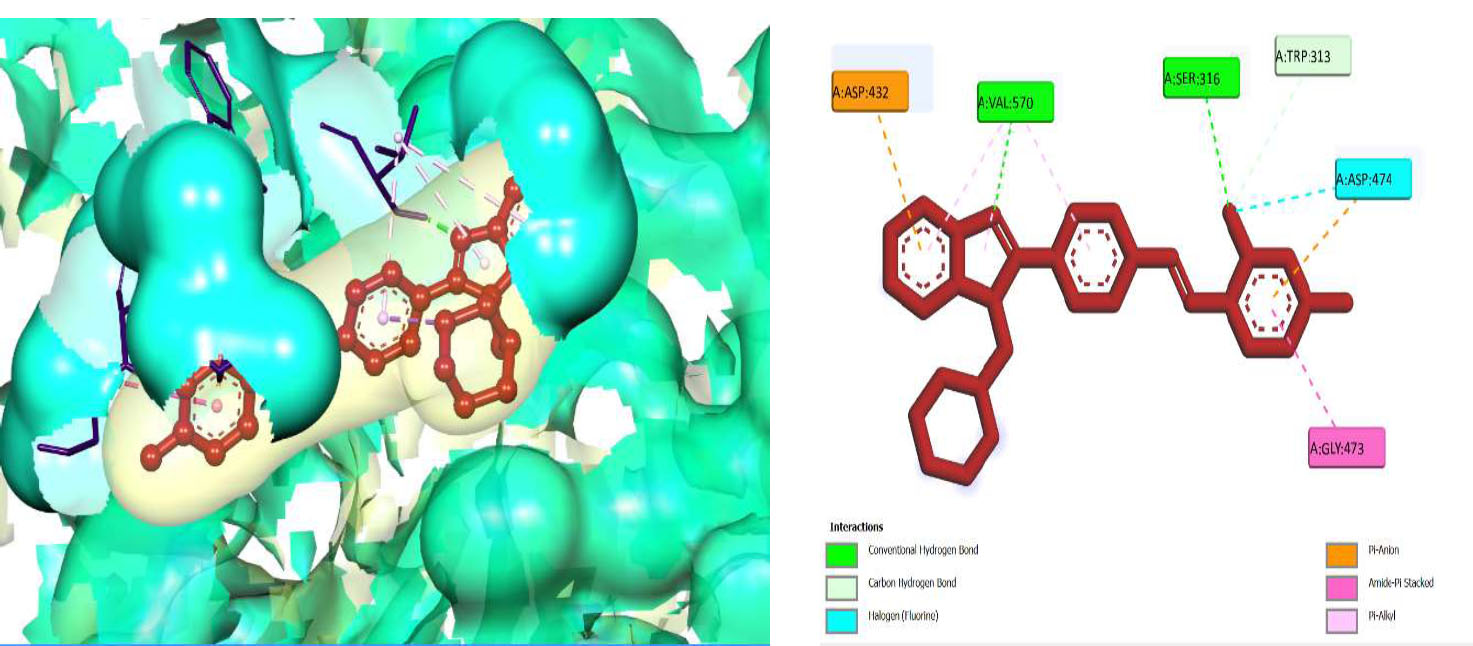
The 3D docked pose and 2D dimension of tested compound 3b on glucosamine-6-phosphate synthase (PDB ID: 1JXA).
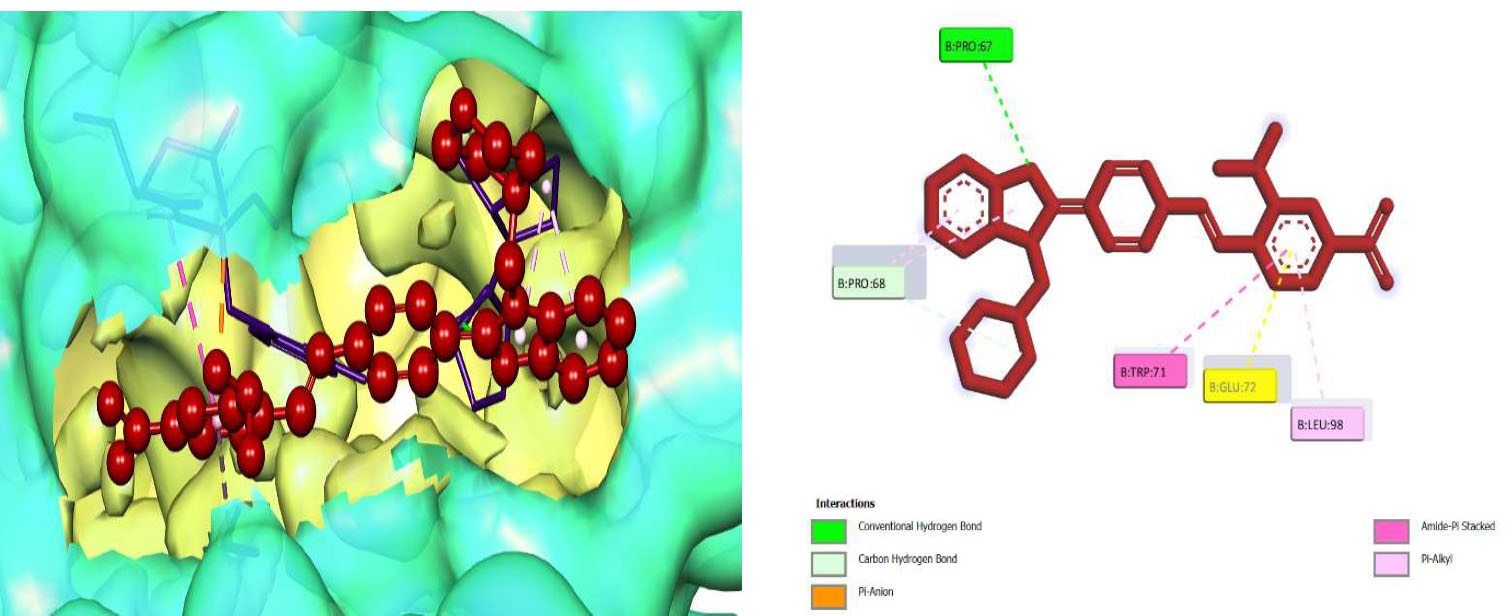
The 3D docked pose and the 2D dimension of tested compound 3f on D-Alanine-D-Alanine Ligase with ATP from Thermus thermophius (PDB ID: 2ZDQ).
3.3. Physicochemical Properties, Pharmacokinetics, and Toxicity Prediction
In the drug discovery process, prediction of the ADMET profile is essential. Due to this, more than 60% of drug failures were attained in clinical trials, which reflects rapidly increasing expense for novel drug development [58, 59]. Due to this reason, prediction of pharmacokinetic profile plays a key role in high throughput screening. These lead optimization efforts can enhance the desirable properties of compounds and eradicate excessive cost and time wastage.
The synthesized compounds were analyzed for ADMET properties by the Qikprop module, in which 3D ligand structures were prepared from the Ligprep tool in Maestro, and its findings were reported (Table 3). The synthesized hybrids afforded excellent scores on oral gastrointestinal absorption and permeable properties of Caco-2 cells on the prediction of percentage human oral absorption and QPPCaco-2, respectively. The findings revealed that compounds 3a, 3b, 3d, 3e, 3g, 3h, 3j, 3k, 3m, and 3n produced 100% oral absorption (except 3d its value is 88.83%) and more than 1200apparent Caco2-permeability (nm/sec) were reported. These results clearly depicted that out of fifteen compounds, except 3f (55.46% HOA and 23.42 Caco-2 permeability) afforded excellent oral absorption and permeability. Being an orally active molecule that should not violate more than two for the rule of five, the findings indicated that most of the tested compounds produced zero violations, except 3a (1 violation), 3d (2 violation), and 3f (1 violation) were reported [61].
Overall, the solubility of a drug should enhance its bioavailability by initiating absorption, distribution, and metabolism. In that point of view, the aqueous solubility of tested compounds was predicted by QPlogs, which afforded the values in the permitted range (-3.27 to –6.00). The permeability of the blood-brain barrier (BBB) was achieved on lipid-soluble drugs, but polar drugs did not cross the blood-brain barrier for access to the central nervous system. The two important parameters employed for the prediction of BBB permeability were blood-brain partition coefficients (QPlogB/B) and CNS activity. The findings showed that compound 3f was found to be inactive (-2), and existing molecules were active (Table 3), which may cause a chance for CNS side effects. Comparatively blood partition coefficients for all tested compounds were within the desirable limit, as -1.73 to 0.66 were reported. The drug moiety binding towards the serum albumin could be an effective key principle for the estimation of dose level for that drug. In that concept, the prediction of human serum albumin binding was estimated (QPlogkhsa), and its findings showed that all tested compounds existed in the expected range (-1.5 to 1.5). Along with that, skin permeation was also predicted (QPlogkp), affording excellent results for all compounds with their desirable range (-1.63 to -3.75).
Madin-Darby canine kidney (MDCK) monolayer predictions were employed for the determination of passive permeability [62]. These MDCK cells were utilized to study the drug efflux (P-glycoprotein) and its active transport [63]. The P-glycoprotein was known to be a recognized efflux transporter that existed commonly in the intestine, brain, and kidney. As a consequence, ligand 3a of our calculated apparent MDCK cell permeability could be considered a good model for the BBB (3796.17), and 3f was the least one (9.46).
The prediction of the physicochemical properties of synthesized hybrids should be necessary in the drug discovery process for enhancing the biological activity of those drug moieties [64]. In this aspect, the optimum values of descriptors like molecular weight, hydrogen bond acceptors, donor, rotational bonds and partition coefficient were predicted (Table 4). The findings showed that molecular weight was in the desirable range of 396 to 554, and the maximum number of hydrogen bond acceptors and donors of tested compounds were 8 and 2, respectively (Table 4). The number of rotational bonds was observed as less than 8 and polar surface area existed within limits (39.16 to 128.19).
| Code | QPlogS | QPPCaco | QPlogBB | CNS | QPPMDCK | QPlogKp | #metab | QPlogKhsa | Percent HOA | Rule of Five |
|---|---|---|---|---|---|---|---|---|---|---|
| 3a | -5.77 | 1263.8 | 0.66 | 2 | 3796.17 | -2.27 | 2 | 0.69 | 100 | 1 |
| 3b | -4.76 | 1268.85 | 0.54 | 2 | 1916.84 | -2.2 | 2 | 0.51 | 100 | 0 |
| 3c | -4.12 | 167.93 | -0.72 | 0 | 79.56 | -3.75 | 4 | 0.34 | 85.72 | 0 |
| 3d | -6.00 | 1264.09 | 0.68 | 2 | 4379.79 | -2.28 | 2 | 0.74 | 88.83 | 2 |
| 3e | -4.50 | 1235.65 | 0.2 | 1 | 687.95 | -2.18 | 4 | 0.38 | 100 | 0 |
| 3f | -4.08 | 23.42 | -1.73 | -2 | 9.46 | -5.53 | 4 | 0.21 | 55.46 | 1 |
| 3g | -4.21 | 1270.35 | 0.35 | 1 | 708.86 | -1.96 | 2 | 0.43 | 100 | 0 |
| 3h | -4.62 | 1330.7 | 0.24 | 1 | 745.33 | -1.63 | 2 | 0.58 | 100 | 0 |
| 3i | -3.27 | 669.51 | 0.06 | 1 | 354.73 | -2.63 | 4 | -0.01 | 96.92 | 0 |
| 3j | -4.98 | 1270.75 | 0.52 | 2 | 1747.7 | -2.13 | 2 | 0.56 | 100 | 0 |
| 3k | -4.59 | 1270.85 | 0.46 | 2 | 1279.64 | -2.09 | 2 | 0.47 | 100 | 0 |
| 3l | -4.47 | 383.32 | -0.28 | 1 | 194.14 | -3.02 | 3 | 0.49 | 95.79 | 0 |
| 3m | -5.10 | 1271.93 | 0.53 | 2 | 1879.59 | -2.13 | 2 | 0.58 | 100 | 0 |
| 3n | -4.26 | 1270.35 | 0.28 | 1 | 708.86 | -2.07 | 3 | 0.38 | 100 | 0 |
| 3o | -3.83 | 294.65 | -0.38 | 1 | 146.09 | -3.28 | 3 | 0.29 | 93.07 | 0 |
| Code | PSA | mol MW | SASA | FOSA | FISA | PISA | WPSA | HBD | HBA | QPlogPoct | QPlogPw | #rotor |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3a | 39.51 | 465.38 | 752.13 | 191.24 | 30.70 | 396.71 | 133.48 | 0 | 6.2 | 19.65 | 9.49 | 5 |
| 3b | 38.26 | 432.47 | 711.31 | 186.82 | 30.52 | 415.01 | 78.97 | 0 | 6.2 | 18.77 | 9.49 | 5 |
| 3c | 80.63 | 428.49 | 716.30 | 186.78 | 123.13 | 406.38 | 0.00 | 2 | 7.7 | 22.43 | 14.01 | 7 |
| 3d | 39.51 | 554.28 | 761.39 | 190.59 | 30.69 | 395.31 | 144.80 | 0 | 6.2 | 19.92 | 9.50 | 5 |
| 3e | 55.46 | 456.54 | 774.25 | 368.41 | 31.73 | 374.11 | 0.00 | 0 | 7.7 | 20.00 | 10.38 | 7 |
| 3f | 128.19 | 486.49 | 766.12 | 181.53 | 213.35 | 371.24 | 0.00 | 0 | 8.2 | 23.03 | 12.07 | 7 |
| 3g | 39.16 | 396.49 | 700.42 | 186.44 | 30.46 | 483.52 | 0.00 | 0 | 6.2 | 18.30 | 9.91 | 5 |
| 3h | 38.28 | 422.53 | 739.30 | 199.15 | 28.34 | 511.81 | 0.00 | 0 | 6.2 | 18.98 | 9.74 | 7 |
| 3i | 52.16 | 397.48 | 692.30 | 186.82 | 59.80 | 445.68 | 0.00 | 0 | 7.7 | 19.16 | 11.31 | 5 |
| 3j | 39.16 | 430.94 | 724.51 | 186.42 | 30.45 | 436.12 | 71.52 | 0 | 6.2 | 19.00 | 9.67 | 5 |
| 3k | 39.16 | 414.48 | 709.45 | 186.42 | 30.45 | 445.79 | 46.80 | 0 | 6.2 | 18.55 | 9.69 | 5 |
| 3l | 61.82 | 412.49 | 713.00 | 186.44 | 85.34 | 441.22 | 0.00 | 1 | 6.95 | 20.49 | 12.00 | 6 |
| 3m | 39.14 | 475.39 | 729.79 | 186.41 | 30.41 | 435.78 | 77.20 | 0 | 6.2 | 19.14 | 9.68 | 5 |
| 3n | 47.63 | 426.52 | 732.38 | 276.59 | 30.46 | 425.32 | 0.00 | 0 | 6.95 | 19.04 | 10.10 | 6 |
| 3o | 79.38 | 441.49 | 716.70 | 186.44 | 97.39 | 432.87 | 0.00 | 0 | 7.2 | 20.34 | 10.82 | 6 |
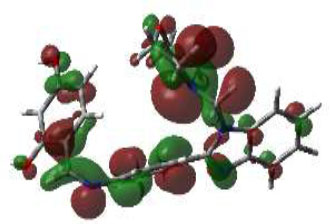
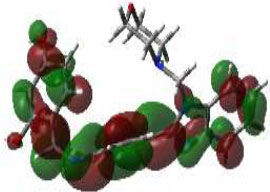
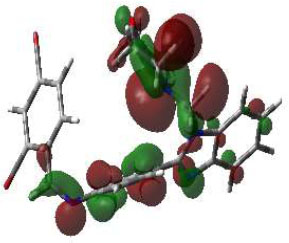
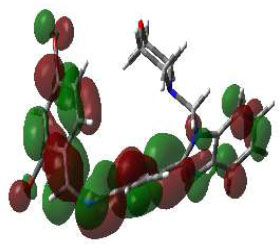
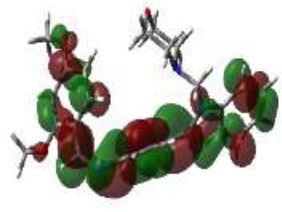
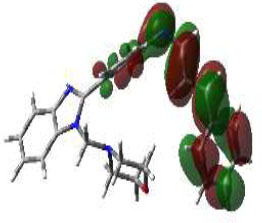
In bioactivities, the frontier-orbital energies of a compound play a vital role, in which, EHOMO is associated with electron-donating properties (nucleophilic attack) and ELUMO is linked to electron-accepting properties (electrophilic attack).
Based on this statement, EHOMO energy values were enhanced by electron-donating groups, and, on another hand, ELUMO energy values declined due to revert performance. The Density Functional Theory (DFT) study results give us important information about the compounds' electronic properties and possible bioactivity (3a–3o), as mentioned in Table 5. The difference between EHOMO and ELUMO (ΔE = ELUMO-EHOMO) represents the chemical reactivity and stability of the molecule. From this perspective, we observed a notable reduction in the energy gap between the high-occupied and lower-unoccupied molecular orbitals in all synthesized compounds. The results showed that the titled compounds had significant EHOMO energy levels that fell within the N-phenyl group on the parent ring. On the other hand, the ELUMO energy levels fell within (1H-benzimidazol-2-yl) benzenamine derivatives. The DFT study suggests that adding an electron-donating group to the parent benzimidazole core can raise the EHOMO energy value and lower the ELUMO energy value. These modifications improve the stability and reactivity of these compounds, which potentially enhances their bioactivity [65].
3.4. Antimicrobial Activity
The synthesized compounds were evaluated for anti-microbial activity by agar diffusion method on both bacterial and fungal strains in which zones of inhibition were measured [66]. Among the synthesized motifs (3a-3o), the compounds 3b, 3d, 3e, and 3n afforded high potential results with the zone of inhibition in the range of 11 to 24 mm, which are mentioned in Table 5. Among these four potential compounds, 3b i.e., [(N-(2,4-difluorobenzylidene)-4-(1-(morpholinomethyl)-1H-benzimidazol-2-yl) benzenamine] with difluoro substitution on ortho and para position of benzylidene group afforded higher percentage inhibition on three gram-positive bacterial strains (Staphylococcus aureus ATCC-6523 [67], Streptomyces albus ATCC-21838 [68], and Micrococcus luteus ATCC-4698), three gram-negative strains (Klebsiella pneumoniae ATCC-33495, Pseudomonas aeruginosa ATCC-BAA3211, and Escherichia coli ATCC-25922) [69], and two fungal strains (Monascus ruber ATCC-20542 and Candida albicans ATCC-10231 [70]) in the range of 91 to 96%. Next to that, the compound 3d i.e., [N-(2,4-dibromobenzylidene)-4-(1-(morpholino-methyl)-1H-benzimidazol-2-yl) benzenamine] with dibromo substitution on ortho and para position of benzylidene group produced potential inhibition in the range of 85% to 96% on all strains, except S.aureus in which results were moderate in their activity. Compound 3e [N-(2,4-dimethoxybenzylidene)-4-(1-(morpholinomethyl)-1H-benzimidazol-2-yl)benzenamine], with dimethoxy substitution at the ortho and para positions of the benzylidene group, exhibited more than 80% inhibition in bacterial strains. In contrast, it showed moderate activity in fungal strains, with inhibition ranging from 55% to 74%.
|
Compound Name |
HOMO | EHOMO (ev) | LUMO | ELUMO (ev) | Energy Gap (Δev) |
|---|---|---|---|---|---|
| 3a |
 |
-8.2785 |
 |
-5.6270 | 2.6515 |
| 3b |
 |
-8.7792 |
 |
-5.6725 | 3.1067 |
| 3c |
 |
-8.7729 |
 |
-5.6714 | 3.1016 |
| 3d |
 |
-8.7784 |
 |
-5.7824 | 2.9959 |
| 3e |
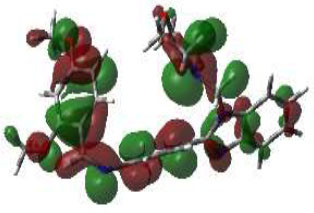 |
-8.7433 |
 |
-5.6597 | 3.0836 |
| 3f |
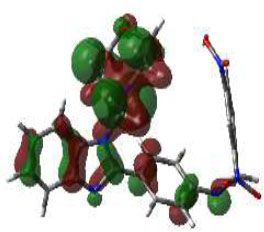 |
-8.8320 |
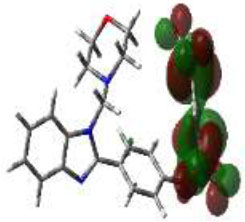 |
-6.2657 | 2.5663 |
| 3g |
 |
-8.8165 |
 |
-5.7955 | 3.0210 |
| 3h |
 |
-8.7523 |
 |
-6.2036 | 2.5486 |
| 3i |
 |
-8.7887 |
|
-6.0347 | 2.7541 |
| 3j |
 |
-8.3343 |
 |
-5.7051 | 2.6292 |
| 3k |
 |
-8.7830 |
 |
-5.7109 | 3.0722 |
| 3l |
 |
-8.7822 |
 |
-5.7051 | 3.0771 |
| 3m |
 |
-8.7838 |
 |
-5.8687 | 2.9152 |
| 3n |
 |
-8.7890 |
 |
-5.5381 | 3.2509 |
| 3o |
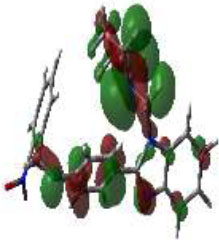 |
-8.7721 |
 |
-6.0695 | 2.7026 |
The compound 3n with para-methoxy substitution on benzylidene group initiated higher potency in percentage inhibition in the range of 84% to 96% except for K.pneumoniae, which produced moderate activity (73%), as shown in Table 6.
The broth dilution method was determined for minimum inhibitory concentration for synthesized compounds [71]. Here, the compounds 3b, 3d, 3e, 3k, and 3n afforded significant MIC in all strains, as shown in Table 7. Compound 3f [N-(2,4-dinitrobenzylidene)-4-(1-(morpholinomethyl)-1H-benzimidazol-2-yl)benzenamine], with dinitro substitution at the ortho and para positions of the benzylidene group, did not show any significant MIC against the fungal strains. In contrast, when compared to the reference drug gatifloxacin [72], compounds 3b, 3e, and 3n exhibited significant MIC activity against the S. albus strain. Additionally, clotrimazole (antifungal agent) showed less potency than compounds 3b, 3d, 3k, and 3n.
| Compound Code | Gram +ve Strains | Gram -ve Strains | Fungal Strains | |||||
|---|---|---|---|---|---|---|---|---|
| S.aureus | S.albus | M. luteus | K.pneumoniae | P. aeruginosa | E. coli | M.ruber | C.albicans | |
|
ZI in mm (PI in %) |
ZI in mm (PI in %) |
ZI in mm (PI in %) |
ZI in mm (PI in %) |
ZI in mm (PI in %) |
ZI in mm (PI in %) |
ZI in mm (PI in %) |
ZI in mm (PI in %) |
|
| 3a |
20±0.28 (80%) |
17±0.25 (71%) |
16±0.17 (70%) |
23±0.20 (88%) |
20±0.24 (83%) |
21±0.19 (84%) |
14±0.17 (70%) |
- |
| 3b |
24±0.25 (96%) |
22±0.28 (92%) |
21±0.22 (91%) |
24±0.18 (92%) |
23±0.21 (96%) |
24±0.15 (96%) |
19±0.15 (95%) |
18±0.19 (95%) |
| 3c | 14±0.19 (56%) |
- | 15±0.25 (65%) |
20±0.16 (77%) |
18±0.17 (75%) |
- | 12±0.11 (60%) |
10±0.15 (53%) |
| 3d | 17±0.23 (68%) |
23±0.19 (96%) |
17±0.19 (74%) |
22±0.24 (85%) |
22±0.15 (92%) |
23±0.16 (92%) |
17±0.19 (85%) |
17±0.16 (89%) |
| 3e |
23±0.14 (92%) |
21±0.27 (88%) |
19±0.24 (83%) |
23±0.11 (88%) |
19±0.22 (79%) |
17±0.12 (68%) |
11±0.22 (55%) |
14±0.11 (74%) |
| 3f | 7±0.29 (28%) |
9±0.17 (38%) |
- | 11±0.12 (42%) |
10±0.19 (42%) |
- | - | - |
| 3g |
9±0.25 (36%) |
- | - | 12±0.25 (46%) |
9±0.12 (38%) |
- | - |
6±0.20 (32%) |
| 3h |
11±0.27 (44%) |
- | 8±0.21 (35%) |
13±0.19 (50%) |
11±0.21 (46%) |
8±0.14 (32%) |
- |
7±0.18 (37%) |
| 3i |
10±0.22 (40%) |
19±0.22 (79%) |
12±0.13 (52%) |
- | 10±0.22 (42%) |
18±0.14 (72%) |
- | 12±0.13 (63%) |
| 3j |
19±0.20 (76%) |
16±0.15 (67%) |
16±0.15 (70%) |
15±0.20 (58%) |
- | 12±0.18 (48%) |
10±0.17 (50%) |
8±0.14 (42%) |
| 3k |
22±0.19 (88%) |
20±0.17 (83%) |
18±0.22 (78%) |
18±0.24 (69%) |
18±0.17 (75%) |
22±0.11 (88%) |
16±0.20 (80%) |
15±0.17 (79%) |
| 3l |
21±0.24 (84%) |
18±0.20 (75%) |
14±0.18 (61%) |
17±0.18 (65%) |
17±0.23 (71%) |
11±0.21 (44%) |
9±0.14 (45%) |
9±0.16 (47%) |
| 3m |
15±0.26 (60%) |
10±0.25 (42%) |
15±0.24 (65%) |
- | 15±0.26 (63%) |
19±0.13 (76%) |
15±0.23 (75%) |
13 ±0.12 (68%) |
| 3n |
22±0.25 (88%) |
23±0.19 (96%) |
20±0.26 (87%) |
19±0.16 (73%) |
21±0.14 (88%) |
22±0.13 (88%) |
17±0.21 (85%) |
16 (84%) |
| 3o | - | 8±0.13 (33%) |
9±0.21 (39%) |
10±0.25 (38%) |
- | - | - | - |
| Standard | 25±0.22* (100%) |
24±0.16* (100%) |
23±0.29* (100%) |
26±0.15* (100%) |
24±0.25* (100%) |
25±0.09* (100%) |
20±0.13** (100%) |
19±0.23** (100%) |
| DMSO | - | - | - | - | - | - | - | - |
| Compound Code | Gram +ve Strains | Gram -ve Strains | Fungal Strains | |||||
|---|---|---|---|---|---|---|---|---|
| S.aureus | S.albus | M. luteus | K.pneumoniae | P. aeruginosa | E. coli | M.ruber | C.albicans | |
| (µg/mL) | (µg/mL) | (µg/mL) | (µg/mL) | (µg/mL) | (µg/mL) | (µg/mL) | (µg/mL) | |
| 3a | 7.8 | 15.6 | 1.9 | 0.95 | 0.95 | 1.9 | 31.3 | NI |
| 3b | 0.5 | 3.9 | 0.95 | 0.5 | 0.23 | 0.5 | 7.8 | 3.9 |
| 3c | 31.3 | NI | 3.9 | 1.9 | 1.9 | NI | NI | 31.3 |
| 3d | 15.6 | 7.8 | 1.9 | 0.95 | 0.5 | 0.95 | 15.63 | 7.8 |
| 3e | 0.95 | 7.8 | 0.95 | 0.95 | 1.9 | 7.8 | 31.3 | 15.6 |
| 3f | NI | 31.3 | NI | 7.8 | 7.8 | NI | NI | NI |
| 3g | 31.3 | NI | NI | 3.9 | 15.6 | NI | NI | 31.3 |
| 3h | 15.6 | NI | 15.6 | 7.8 | 7.8 | 31.3 | NI | NI |
| 3i | NI | 15.6 | 7.8 | NI | 15.6 | 3.9 | NI | 31.3 |
| 3j | 15.6 | 31.3 | 1.9 | 7.8 | NI | 15.6 | 31.3 | 15.6 |
| 3k | 1.9 | 15.6 | 0.95 | 3.9 | 1.9 | 1.9 | 15.63 | 15.6 |
| 3l | 3.9 | 31.3 | 7.8 | 3.9 | 1.9 | 15.6 | NI | NI |
| 3m | 15.6 | 15.6 | 3.9 | NI | 3.9 | 3.9 | 31.3 | 31.3 |
| 3n | 1.9 | 7.8 | 0.5 | 1.9 | 0.5 | 1.9 | 7.8 | 7.8 |
| 3o | NI | 31.3 | 15.6 | 15.6 | NI | NI | NI | NI |
| Standard | 0.23* | 15.6* | 0.5* | 0.23* | 0.46* | 0.95* | 31.25** | 15.63** |
Recent literature has correlated these results with the presence of electron-withdrawing groups, leading to enhanced activity. This enhances lipophilicity and electronic interactions, facilitating better bacterial membrane penetration and target binding. This aligns with the observed efficacy of benzimidazole derivatives substituted with morpholine and benzylidene groups in this study, further supporting the role of EWGs in enhancing antimicrobial activity [73].
Many antimicrobial agents act by targeting essential biological processes in microbial cells, such as protein synthesis, DNA replication, or cell membrane integrity. However, these pathways often share similarities with those in human cells, particularly in rapidly dividing human cells, such as cancer cells. As a result, unintended interactions with human systems may lead to cytotoxic effects. For instance, antimicrobial compounds designed to inhibit bacterial ribosomes can inadvertently interfere with mitochondrial ribosomes in human cells due to their structural resemblance. Similarly, amphipathic antimicrobial agents, such as polymyxins, disrupt bacterial membranes by interacting with their lipid components. However, these compounds may also compromise the integrity of human cell membranes, resulting in potential toxicity. To develop safer antimicrobial agents, it is crucial to achieve a balance between high antimicrobial efficacy and minimal cytotoxicity in human cells. Often, we can achieve this balance by optimizing the chemical structure of the compounds, which enhances microbial selectivity and reduces off-target effects on human systems. In this point of view, assessment of pharmacokinetic and toxicity parameters denoted that all synthesized compounds produced significant results except for 3f, containing the 2,4-dinitro group and exhibiting CNS toxicity and carcinogenicity.
Benzimidazole compounds that combine Schiff bases and morpholine structures show great promise for treating infections and cancer due to their flexible structure and ability to impact biological processes. Making specific changes to the structure of benzimidazole, like adding halogen or methoxy groups, greatly boosts its strong antimicrobial and anticancer effects. These modifications improve how well it targets harmful cells and lower its toxicity to healthy cells [74]. Schiff bases are simple to synthesize, and they can easily change their chemical properties. They have strong antimicrobial effects because they can form complexes with metal ions, which disrupts the function of microbial cells and increases lipophilicity [75, 76]. Morpholine-substituted derivatives are helpful in fighting germs by damaging cell membranes and inhibiting specific key enzyme pathways. The combination of these pharmacophores creates a synergistic effect, as studies have demonstrated that Schiff base and morpholine groups amplify the antimicrobial potential of benzimidazole scaffolds while maintaining favorable safety profiles [73]. These findings underscore the importance of optimizing the physicochemical and pharmacokinetic properties of these hybrid compounds to achieve superior antimicrobial activity with minimal cytotoxicity.
3.5. Structural Activity Relationship
Based on the results of the docking study, ADMET and biological evaluations on synthesized benzimidazole derivatives (3a-3o) substituted with morpholine and benzylidene groups were enhanced, and the antimicrobial effect was observed in all synthesized motifs. Particularly, the
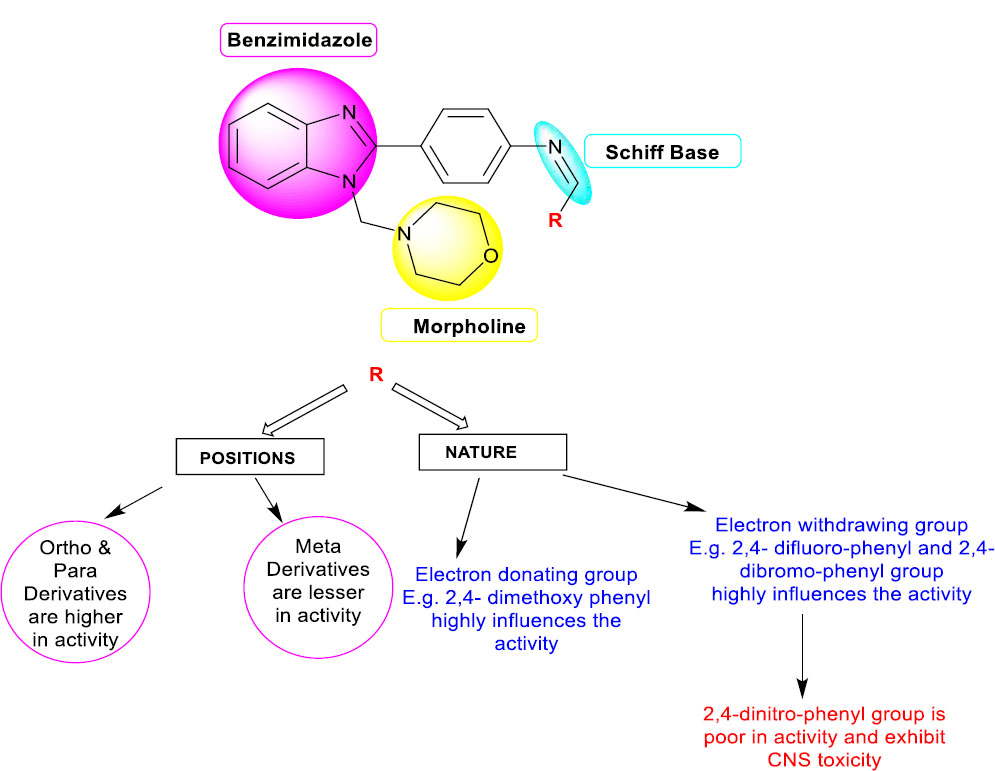
Structural activity relationship of synthesized benzimidazole derivatives (3a-3o).
compounds 3b (2,4-difluoro substitution), 3d (2,4-dibromo substitution), 3e (2,4-dimethoxy substitution), and 3n (4-methoxy substitution) produced enhanced effects compared to their respective standards [77]. However, based on the results of pharmacokinetic and physicochemical parameters, compound 3d with dibromo substitution produced average results and possessed 2 violations in the Lipinski rule. On another hand, compound 3f with 2,4-dinitro substitution produced CNS toxicity, and 50% human oral absorption was observed [73]. Due to that reason, the compound 3f (Nitro substituted) was devoid of an antimicrobial effect. In-vitro assays were reflected in the zone of inhibition, and MIC was observed. The hydrogen bond interactions were also favourable for producing lower binding energy on the compounds 3b, 3e, and 3n.
Based on recent literature, benzimidazole analogues substituted with Schiff bases and morpholine have demonstrated an enhanced broad-spectrum activity against both bacterial and fungal pathogens, which is well-supported by this study [78, 79]. Overall, the designed pharmacophore as benzimidazole derivatives substituted with Schiff base and morpholine showed improved anti-microbial activity than the benzimidazole motif (Fig. 5).
CONCLUSION
The study concluded that designed and synthesized benzimidazole derivatives, with morpholine and Schiff base motifs, enhanced antimicrobial activity against both bacterial and fungal pathogens. The molecular docking studies were consistent with the results of in-vitro assays, further supporting the hypothesis of this research.
Among the fifteen compounds, 3b, 3e, and 3n produced noteworthy results in antimicrobial screening compared to their reference drugs. Overall, the activity of synthesized motifs was based upon the three pharmacophores, benzimidazole, morpholine, and Schiff bases, with an electron-withdrawing group (fluoro and bromo substitution) and electron donating group (methoxy substitution) on the benzene ring.
The complete binding pattern of synthesized benzimidazoles was further explored by altering substituent groups on the pharmacophore, which should improve the inhibitory action of targeted enzymes. The pharmacokinetics and physicochemical properties of these synthesized hybrids provide insights into the development of antimicrobial agents. Further studies are needed to explore molecular mechanisms through cell line studies, and evaluation of toxicological studies will be helpful in identifying potential antimicrobial candidates.
AUTHORS’ CONTRIBUTIONS
It is hereby acknowledged that all authors have accepted responsibility for the manuscript's content and consented to its submission. They have meticulously reviewed all results and unanimously approved the final version of the manuscript.
LIST OF ABBREVIATIONS
| E. coli | = Escherichia coli |
| MDCK | = Madin-Darby Canine Kidney |
| DFT | = Density Functional Theory |
| BBB | = Blood-brain Barrier |
AVAILABILITY OF DATA AND MATERIALS
The data supporting the findings of this study are available within the article and its supplementary materials.
ACKNOWLEDGEMENTS
Declared none.

