All published articles of this journal are available on ScienceDirect.

The Role of the BCL2 Family in Targeted Anticancer Response as Apoptotic Mediators
Abstract
The process known as “programmed cell death,” or apoptosis, is mediated by caspases and regulates tissue homeostasis. There are two pathways by which this process occurs: the intrinsic mechanism, also known as the mitochondrial pathway, and the extrinsic pathway, which is mediated by death receptors. The intrinsic path, which initiates cell death by permeabilizing the mitochondrial membrane and releasing components that induce apoptosis, is regulated by the BCL-2 protein family, which is encoded by the BCL-2 gene. These family proteins' four BCL-2 homology domains (BH1-4) are essential to their operation, and their shared BH domains enable smoother interactions among members of the same family and can also be used as markers of pro- or anti-apoptotic activity. Cell death may be delayed because of BCL-2 overexpression. Several cancers, such as lung, breast, melanoma, and chronic lymphocytic leukemia, as well as Multiple Sclerosis and diabetes, have been linked to changes in BCL-2 expression. This review examines the importance of BCL-2 family interactions for both health and disease, as well as the therapeutic potential to modulate them.
1. INTRODUCTION
The fundamental process of apoptosis, in which cells are programmed to die, maintains equilibrium within the body and eliminates damaged or unneeded cells without causing an immune response or threatening tissues around them [1]. Apoptotic cell death is often triggered by infections, helping eliminate infected cells [2, 3]. Cells compress, chromatin condenses, and the plasma membrane forms blebs as a result of the apoptotic process [4]. The frequency, appearance, and biology of this process set it apart from other forms of cell death [5], and its dysregulation can lead to cancer and neurological, autoimmune, cardiovascular, and infectious diseases [6].
There are two primary pathways that regulate apoptosis: the intrinsic and extrinsic pathways [7]. Transmembrane proteins of the death receptor family, including TNF-R1, CD95, TRAIL-R1, TRAIL-R2, DR3, and DR6, bind to their corresponding ligands (TNF, CD95L, TRAIL, and TL1A) to initiate the extrinsic pathway, which leads to the formation of the signals that cause death and caspase-8 activation [8]. Then, caspase-8 can either generate the intrinsic (mitochondrial) apoptosis pathway by proteolytically cleaving Bid into tBid or cleave caspase-3 or other effector caspases [8]. The BCL-2 protein family, which includes both pro- and antiapoptotic members that regulate cell death, is essential to this process [9, 10].
When caspase-8 is activated, tBid travels to the mitochondrial membrane and interacts with other members of the BCL-2 family. This causes the outer membrane of the mitochondria to open, allowing proteins, such as cytochrome c, to be released into the cytosol [8, 10]. At this point, the BCL-2 protein family is essential in the cell's decision to activate this pathway and commit suicide [9, 10]. In conclusion, apoptosis is an essential biological process that is controlled by two different pathways and contributes to the removal of damaged or unneeded cells as well as to the maintenance of organ homeostasis. Apoptosis dysregulation can result in a number of illnesses, and the BCL-2 protein family is essential for controlling cell death [11, 12].
The BCL-2 (B-cell lymphoma-2) family comprises key mediators of apoptosis and was initially identified as a proto-oncogene in lymphoma cells [13]. The BCL-2 gene was discovered at the t(14;18) chromosomal translocation breakpoint, a rearrangement that brings the BCL-2 locus on chromosome 18q21 under the influence of strong immunoglobulin heavy chain (IGH) enhancers on chromosome 14q32, leading to dysregulated BCL-2 expression [14, 15]. The term BCL-2 family refers to a group of proteins sharing BCL-2 homology (BH) domains, which are critical for mediating protein-protein interactions and determining pro- or anti-apoptotic function [16].
Based on their structure and function, BCL-2 family proteins are classified into three subgroups: (1) antiapoptotic (prosurvival) proteins, (2) proapoptotic pore-forming proteins, and (3) proapoptotic BH3-only proteins [17]. The antiapoptotic subgroup includes BCL-2, BCL-XL, BCL-W, BCL-B, MCL-1, and BFL-1/A1, all of which inhibit apoptosis and share the characteristic N-terminal BH4 domain, a hallmark of prosurvival proteins [18]. In addition to regulating apoptosis, BCL-2 and BCL-XL have been shown to influence proliferation, autophagy, differentiation, DNA repair, tumor growth, and angiogenesis, largely through the activity of their BH4 domain [19].
Both proapoptotic and antiapoptotic members contain up to four BH domains (BH1–BH4). These domains form a conserved structural motif, consisting of a central hydrophobic core composed of two α-helices and surrounded by six to seven amphipathic α-helices. This configuration creates a hydrophobic BH3-binding cleft, which acts as a receptor for the BH3 domain of proapoptotic proteins [15, 20]. Antiapoptotic proteins suppress apoptosis by binding and neutralizing proapoptotic counterparts through these conserved BH domains [21].
The proapoptotic BH3-only proteins BID, BIM, PUMA, BIK, BAD, BMF, NOXA, and HRK share a conserved BH3 domain consisting of approximately 9–16 amino acids, which is essential for their apoptotic function [22, 23]. These proteins are crucial initiators of apoptosis, triggered by both developmental cues and cellular stress. Based on their function, BH3-only proteins are subdivided into two classes: activators and sensitizers [24]. Activator proteins, such as BID, BIM, PUMA, and NOXA, directly bind to the hydrophobic groove of BAX and BAK, inducing conformational changes that lead to their activation. In contrast, sensitizer proteins BAD, BIK, BMF, and HRK promote apoptosis indirectly by binding to antiapoptotic BCL-2 family members, thereby displacing activator proteins and allowing them to engage BAX/BAK [25-27]. Some BCL-2 family proteins, however, do not fit neatly into existing classification frameworks due to ambiguous functions and structural variations in their BH domains. For instance, BCL-2 family kin (BFK) refers to a poorly characterized member of the family that contains both BH2 and BH3 domains, yet exhibits limited proapoptotic activity upon forced expression. BFK is predominantly expressed in the human gastrointestinal tract, and its expression is significantly downregulated in cancers derived from various digestive organs, suggesting a potential, albeit limited, role in tumor suppression [20].
This research highlights the dual roles of BCL-2 family members as both proapoptotic and antiapoptotic regulators across a variety of biological processes. Special emphasis was placed on the interactions among BCL-2 family proteins, which are critical in determining cellular fate. Furthermore, the study discusses a range of therapeutic agents targeting BCL-2 family proteins, with relevance to the treatment of diseases, such as multiple sclerosis, diabetes, lung cancer, breast cancer, and Parkinson’s disease.
1.1. Search Strategy
A comprehensive literature search was conducted using the Web of Science, PubMed, and ScienceDirect databases. Relevant articles were identified, and all references included in this review were retrieved from these sources. Additionally, one of our previously published articles (2016) [66] directly addresses the central topic of this review and has been included for contextual relevance.
The search strategy focused on studies exploring the deregulation of apoptosis in various human diseases, particularly emphasizing the role of the Bcl-2 family of proteins. Heat-induced apoptosis was considered a model system for investigating protein misfolding and its consequences. The review also examines the significance of Bcl-2 family protein interactions in health and disease, along with their therapeutic potential. The following keywords and search terms were applied during the literature search: Apoptosis, Bcl-2 family proteins, programmed cell death, mitochondrial membrane permeability, and therapeutic agents.
The following cancer types were considered: lung cancer, breast cancer, melanoma, and lymphocytic leukaemia Boolean operators, such as AND and OR, were used to combine search terms appropriately (e.g., “Apoptosis AND Bcl-2 family proteins,” “Apoptosis AND cancer,” “Bcl-2 OR Bax AND therapeutic agents”). Only peer-reviewed English-language articles were included, with no restrictions on publication year to ensure comprehensive coverage of relevant studies.
2. GENOMIC-PROTEOMIC CONTENT
2.1. Location of the BCL-2 Family Gene within the Genome
The BCL-2 family of proteins is central to the regulation of apoptosis. Since the discovery of BCL-2 in the 1980s in association with B-cell lymphoma [16], a broad array of related proteins has been identified. The intrinsic apoptotic pathway is controlled by approximately 20 members of the BCL-2 gene family, which collectively maintain the critical balance between cell survival and programmed cell death [28]. In response to apoptotic stimuli, several BCL-2 family members translocate from the cytoplasm to the mitochondria. Moreover, the BCL-2 gene itself is expressed across various intracellular membranes, including the mitochondria and endoplasmic reticulum, underscoring its fundamental role in cellular homeostasis.
The BCL-2 gene (B-cell lymphoma 2) was originally identified at the t(14;18) chromosomal translocation breakpoint in B-cell follicular lymphomas, where the immunoglobulin heavy chain gene enhancer and promoter on chromosome 14 drive aberrant transcriptional activation of BCL-2 (Fig. 1) [29-31]. This strong association between BCL-2 and the follicular lymphoma translocation provided early evidence that BCL-2 functions as an oncogene [32]. The translocation results in the fusion of the BCL-2 gene on chromosome 18 with regulatory elements of the immunoglobulin heavy chain gene on chromosome 14, leading to overexpression of the BCL-2 protein [21]. Approximately 90% of follicular lymphomas and 30% of diffuse large B-cell lymphomas harbor this characteristic translocation [33]. Members of the BCL-2 family are defined by the presence of conserved BCL-2 homology (BH) domains (Table 1) [29]. Importantly, the oncogenic role of the BCL-2 translocation is not due to enhanced proliferation but rather the inhibition of apoptosis, thus contributing to tumorigenesis by impairing programmed cell death [30].
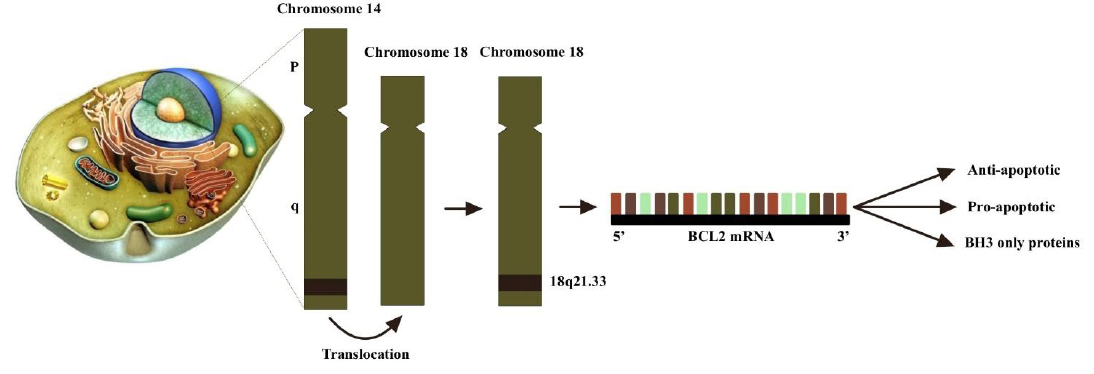
The translocation t(14;18) involving the BCL-2 gene is frequently observed in human B-cell lymphomas, particularly in follicular center B-cell lymphomas. This chromosomal translocation relocates the BCL-2 gene to the 5' immunoglobulin heavy chain (IgH) intron enhancer E on chromosome 14, from its normal position on chromosome 18q21. The BCL-2 family of proteins is categorized into three groups based on their primary functions: (1) pro-apoptotic proteins, including BAX, BAK, and BOK; (2) anti-apoptotic proteins such as BCL-2, BCL-XL, BCL-W, MCL-1, and BFL-1/A1; and (3) BH3-only proteins, which comprise BID, BAD, BIM, BIK, BMF, HRK, NOXA, PUMA, among others. A characteristic feature of all family members is the presence of a BH3 domain-a conserved motif involved in protein-protein interactions within the family. Anti-apoptotic and pore-forming members are multi-BH domain proteins, characterized by containing all four BH domains and a highly conserved tertiary structure that forms a hydrophobic groove capable of binding BH3 domains from other family members.
| Type | Approved Gene Symbol | Approved Gene Name | Aliases | Chromosomal Location | References |
|---|---|---|---|---|---|
| Anti-apoptotic | BCL-2 | BCL-2 apoptosis regulator | BCL-2, PPP1R50 | 18q21.33 | [30, 31] |
| BCL-2L1 | BCL-2-like 1 | BCLX, BCL-2L, BCL-X, BCL-XL, BCL-XS, PPP1R52 | 20q11.21 | [1] | |
| BCL-2L2 | BCL-2 like 2 | KIAA0271, BCL-W, PPP1R51 | 14q11.2 | [34] | |
| MCL1 | MCL1 apoptosis regulator, BCL-2 family member | BCL-2L3, Mcl-1 | 1q21.2 | [35] | |
| BCL-2L10 | BCL-2 like 10 | Diva, Boo, BCL-B | 15q21.2 | [36] | |
| BCL-2A1 | BCL-2-related protein A1 | GRS, BFL1, BCL-2L5, HBPA1 | 15q24.3 | [37] | |
| Pro-apoptotic | BAX | BCL-2-associated X, apoptosis regulator | BCL-2L4 | 19q13.3 q13.4 | [38] |
| BAK1 | BCL-2 antagonist/killer 1 | BCL-2L7, BAK | 6p21.31 | [37, 38] | |
| BOK | BCL-2 family apoptosis regulator BOK | BCL-2L9, BOKL, MGC4631 | 2q37.3 | [36] | |
| BNIP2 | BCL-2 interacting protein 2 | Nip2, BNIP-2 | 15q22.2 | [36] | |
| BH3 only protein | BIK | BCL-2-interacting killer (apoptosis-inducing) | NBK, BBC1 | 22q13.31 | [39] |
| PMAIP1 | Phorbol-12-myristate- 13- acetate-induced protein 1 | NOXA, APR | 18q21 / (18q21.31) | [40, 41] | |
| BBC3 | BCL-2 binding component 3 | JFY1, PUMA | 19q13.3-q13.4 | [42] | |
| HRK | Harakiri, BCL-2 interacting protein (contains only BH3 domain) | DP5 | 12q24.2 | [36] | |
| BMF | BCL-2 modifying factor | - | 15q14 | [43] |
2.2. Structures of BCL-2 Family Members
The three-dimensional structures of BCL-2 family proteins are characterized by six to seven amphipathic α-helices of varying lengths, along with two core predominantly hydrophobic α-helices. These are separated by a long, unstructured loop between the first two helices. Notably, the overall structural fold of BCL-2 proteins closely resembles that of the pore-forming domains found in certain bacterial toxins [28, 44]. BCL-2 family members contain conserved BCL-2 homology (BH) domains, regions of sequence similarity that define their classification [45]. The multi-domain BCL-2 proteins, which possess multiple BH motifs, include both antiapoptotic (e.g., BCL-2, BCL-XL, Bcl-w, Mcl-1, A1, and Bcl-B) and proapoptotic (e.g., BAX and BAK) members. In contrast, the proapoptotic BH3-only proteins, such as Bim, Bad, tBid, Bmf, Bik, Noxa, Puma, and Hrk, contain only the BH3 domain. This BH3 motif, essential for proapoptotic function, is present in nearly all proapoptotic BCL-2 proteins but may be absent in antiapoptotic counterparts (Fig. 2) [1].
The BCL-2 family proteins share four conserved regions known as BCL-2 homology (BH) domains, BH1, BH2, BH3, and BH4, which represent key sites of structural homology among family members and are critical for their function. These domains are essential for mediating protein-protein interactions, including heterodimerization between pro- and anti-apoptotic members, as demonstrated by studies involving domain-specific mutations and deletions [36]. Approximately 44% of the amino acid sequence of BCL-XL (also known as BCL-2L1, BCL-2-like 1) is homologous to BCL-2, and it exists in multiple isoforms beyond the well-characterized BCL-XL and BCL-XS variants [16]. The BH domains in BCL-XL are composed of eight α-helices (α1-α8), which are essential for its tertiary structure. A hydrophobic pocket formed by the BH1–BH3 regions serves as the binding site for the BH3 domain of pro-apoptotic proteins, triggering oligomerization [46]. Although the precise structure of BCL-XS remains unresolved, the loss of both BH1 and BH2 domains would likely result in significant disruption of the hydrophobic binding cleft [16]. Structural studies of BAK and BAX monomers reveal a globular conformation with a hydrophobic core surrounded by eight α-helices [21]. In BAK, the carboxyl-terminal transmembrane helix (helix 9) is constitutively inserted into the mitochondrial outer membrane (MOM). In contrast, BAX is primarily cytoplasmic due to helix 9 being sequestered within the hydrophobic groove formed by the BH3 domain-binding interface [17]. Bid, another pro-apoptotic family member, contains six amphipathic helices surrounding two central, largely hydrophobic α-helices, with the most notable conformational changes occurring in the N-terminal region of the protein [44].
2.3. Gene Expression of BCL-2 Family Protein
A variety of cytotoxic stimuli can harm cells, but BCL-2 expression can stop this damage. Hodgkin's lymphoma (NHL), myeloma, lung, prostate, and breast cancer, melanoma, acute myeloid leukemia (AML), chronic lymphocytic leukemia (CLL), and non-lymphoma all have BCL-2 overexpression [29, 47]. MicroRNAs (miRNAs) control the downregulation/deletion of genes that cause BCL-2, chromosome translocation, and gene amplification. BCL-2 levels may rise due to a variety of processes, including RNA degradation (Table 2) [29].
In CLL, the most prevalent leukemia in humans, BCL-2 overexpression was caused by the absence of regulation of miRNA 15/16 [48]. Differentiating cells have exceptional amounts of BCL-XL expression, which persists throughout neural ontogeny at greater levels. Ablation of the BCL-XL gene, BCL-2l1, causes significant apoptosis in the developing neurological and hematological systems as well as embryonic death by E13.5 [13]. Tumor development is significantly influenced by BCLXL. Previous studies have demonstrated that the BCL-2 L1 gene, which codes for BCLXL, is amplified in many solid tumors [21].
A poor prognosis for patients with human cervical neoplasms and ovarian cancer has been linked to MCL-1 expression. Through a PI3K/Akt-dependent mechanism, IL-6 inhibits cellular apoptosis and encourages the oncogenesis of cervical cancer [33]. Antiapoptotic protein overexpression can result in treatment resistance in addition to lowering normal cell death. Overexpression of BCL-XL and other anti-apoptotic proteins has been associated with therapeutic resistance, as it interferes with the apoptotic pathways targeted by many anticancer agents [49]. In various malignancies, including hepatocellular carcinoma, multiple myeloma, chronic lymphocytic leukemia (CLL), and chronic myeloid leukemia (CML), elevated expression of anti-apoptotic proteins, particularly BCL-XL and Mcl-1, has been correlated with treatment resistance and poor clinical outcomes [33, 47].
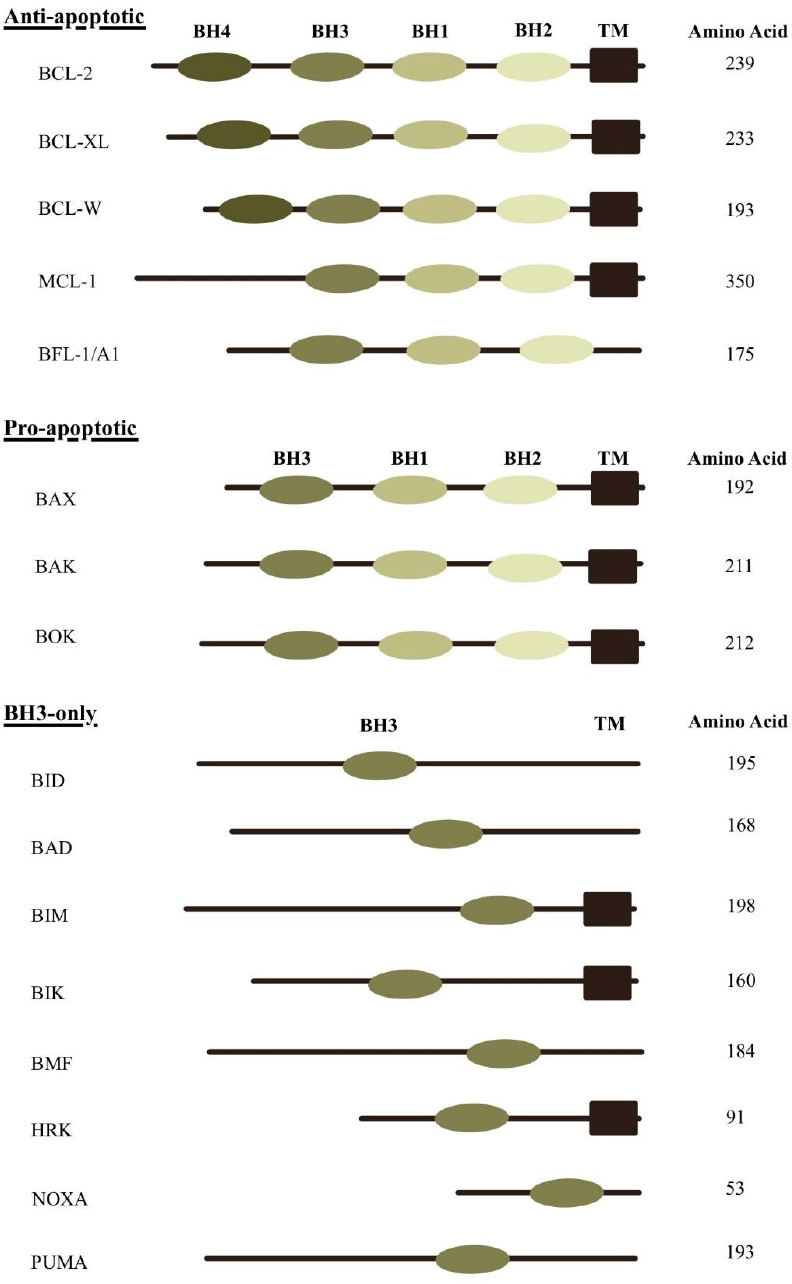
The BCL-2 protein family is categorized into three major groups based on domain structure and function: multidomain pro-apoptotic proteins, multidomain anti-apoptotic proteins, and BH3-only proteins. These proteins are characterized by the presence of conserved BCL-2 homology (BH) domains and, in many cases, a transmembrane (TM) domain that anchors them to intracellular membranes. The pro-apoptotic proteins BAX, BAK, and BOK contain BH1, BH2, and BH3 domains. The anti-apoptotic members, such as BCL-2, BCL-XL, and BCL-W, possess BH1 through BH4 domains, while MCL-1 and BFL-1/A1 include BH1 to BH3. In contrast, BH3-only proteins-including BID, BAD, BIM, BIK, BMF, HRK, NOXA, and PUMA-contain only the BH3 domain, which is essential for initiating apoptosis through interaction with other BCL-2 family members.
| Type | BCL-2 Protein | Subcellular Localization | Function | Reference |
|---|---|---|---|---|
| Anti-apoptotic | BCL-2 | Consistently linked to the MOM and/or ER membrane. | Both apoptosis effectors and BH3-only proteins are inhibited. | [21, 50] |
| BCL-XL | Located in the nuclear envelope, endoplasmic reticulum (ER), and mitochondrial membrane surfaces. | Binds to BAX or BAK to prevent apoptosis. | [17, 51] | |
| MCL-1 | Placement in the outer mitochondrial membrane (OMM) and inner mitochondrial membrane (IMM). | BH3-only proteins and apoptotic effectors are inhibited. It has an impact on cellular health and development in addition to its known pro-survival activities. | [52] | |
| BCL-W | Loosely connected to the mitochondrial membrane and cytosol. | Inhibits BH3-only proteins as well as apoptotic effectors. | [17, 21, 34] | |
| A1/Bfl-1 | Identified in the golgi apparatus, cytosol, nuclear envelope, and ER membranes. | Opposes the activation of caspases and prevents the release of proapoptotic cytochrome c from mitochondria. | [1, 53] | |
| Pro-apoptotic | BAK | Always present on the OMM. | Oligomerizes to create holes in the OMM, which is considered to enhance MOMP. | [54] |
| BAX | Present on the OMM, often present in the cytoplasm as an inactive monomer. | Oligomerizes to create holes in the OMM, which is considered to enhance MOMP. | [54] | |
| BOK | Detected on the ER membranes, nuclear outer membrane, and Golgi apparatus. | Depends on BAX and BAK to promote apoptosis. | [55] | |
| BH3 only protein | BAD | Both the cytosol and the mitochondrial membrane | BAD is referred to as an “indirect” promoter of apoptosis because it causes cell death by inhibiting antiapoptotic proteins. | [56] |
| BIM | Nucleus envelope, endoplasmic reticulum (ER), cytoplasm, and mitochondrial outer membrane (MOM). | In apoptotic induction, it also controls mitochondrial activity and cellular metabolism. | [54, 57] | |
| PUMA | Placement on the mitochondrial outer membrane (MOM), | Crucial p53-dependent apoptosis mediator. Activates BAK and BAX, resulting in MOMP. | [17, 58] | |
| BIK | Mostly found in the ER membrane | Directly impacts BCL-2 and BCL-2-like 1 | [1, 59] |
By increasing the transcriptional factor specificity protein 1's expression, BCLW stimulates tumor invasion and the subsequent generation of matrix metalloproteinase 2 [21]. BCL-2L10 (also called BCL-B or DIVA) is the most important maternally inherited mRNA, according to an analysis of early mRNA expression in early embryos. It is highest in mature eggs but decreases during division [13].
As a result of diverse regulatory mechanisms, BH3-only proteins tend to be underproduced or functionally impaired in both tumor cells and actively proliferating normal cells. While some BH3-only proteins are constitutively expressed, others are transcriptionally induced in response to cellular stressors, such as DNA damage or hypoxia [60]. These observations support the rheostat model, which posits that cell fate is determined by the relative levels of proapoptotic and antiapoptotic BCL-2 family proteins. The discovery of BAK (BCL-2 antagonist/killer), a proapoptotic protein structurally more similar to BCL-2 than to BAX, yet functionally analogous to BAX, further demonstrated that multiple proteins beyond BCL-2 can exert antiapoptotic control [50].
The BAX promoter is broadly active in both normal and malignant tissues and is a direct transcriptional target of the tumor suppressor TP53 [21]. Additionally, BCL-2A1 (also known as A1) was found to be overexpressed in gastric cancer relative to normal tissue, suggesting its potential role in the development of solid tumors [37]. Although PUMA expression is typically low under basal conditions, it can be strongly induced by transcription factors, such as p53, p73, E2F1, and FOXO3a, which may initiate an apoptotic response [61].
3. APOPTOTIC PATHWAY
Cell destruction during eukaryotic development and the preservation of organismal homeostasis are often caused by the evolutionarily conserved cell death mechanism known as apoptosis. By striking a balance between cellular life and death, the BCL-2 protein family, which consists of both pro- and anti-apoptotic members, controls this process [62]. An energy-dependent series of chemical processes makes up the more intricate mechanism of apoptosis [63]. Two distinct mechanisms can cause apoptosis: the extrinsic pathway, which is triggered when death receptors on the cell surface are engaged, and the intrinsic pathway, which is triggered downstream of physiological stressors, such as growth factor withdrawal, ER stress, or DNA damage [64]. Furthermore, a distinct route involving perforin/granzyme-dependent and T-cell-mediated cell death is documented. Granzyme types A and B mediate this pathway [1]. When caspases (aspartate-specific cysteine proteases) are active, these pathways come together. Essential proteins are broken down by these enzymes, which eventually results in the death of cells [65].
Signals inside the cell, such as oncogenic stress, viral infection, or cytotoxic insults that harm DNA or protein molecules, initiate the intrinsic pathway, also known as the mitochondrial pathway. The whole process is regulated by both pro- and anti-apoptotic BCL-2 family members [66]. Important sensors of cellular stress, BH3-only proteins, start the intrinsic apoptosis signaling cascade. By attaching to the BH3 domain, they neutralize the function of anti-apoptotic BCL-2 family members, suppressing their activity and encouraging apoptosis. The apoptotic effectors BAX and BAK become active when the anti-apoptotic BCL-2 protein is suppressed. BAX and BAK may also be directly bound and activated by some BH3-only proteins, such as BIM, tBID (an activated form of BID), and maybe PUMA [65]. Permeation of the Mitochondrial Outer Membrane (MOMP) results from the oligomerization of activated BAX and BAK. Intermembrane proteins, such as cytochrome c, a second mitochondria-derived activator of caspase (SMAC, also called DIABLO), AIF (apoptosis-inducing factor), and Omi, may be released as a result of permeabilization [29, 63, 67]. The apoptosome is then created. Cytochrome c, procaspase-9, deoxyadenosine triphosphate (dATP), and apoptotic protease-activating factor-1 (APAF-1) form the complex known as the apoptosome. Within the apoptosome, procaspase-9 transforms into caspase-9 [68]. After that, the apoptosome triggers caspase-9, which triggers caspase-3 and apoptosis [1]. Cell death is regulated by additional pathways involved in intrinsic apoptosis. Omi inhibits the X-linked inhibitor of apoptosis protein (XIAP), an endogenous inhibitor of caspase function [69]. SMAC is released upon the formation of the apoptosome to suppress the inhibitor of apoptosis protein (IAP), hence permitting apoptosis [58]. Direct nuclear translocation by AIF leads to caspase-independent nuclear modifications [63].
Tumor necrosis factor receptor (TNFR), FAS, TNF-related apoptosis-inducing ligand receptor (TRAILR), and cell surface death receptors all trigger the extrinsic pathway when their corresponding ligands, TNF, FASL, and TRAIL, are stimulated [64]. The DD domain in the adaptor protein FADD (Fas-associated death domain) docks with the DD domain of death receptors. Now, procaspase-8 can be drawn to the death effector domain (DED) in FADD and concentrated on the activated receptor by connecting with the DED domain in the prodomain of caspase-8. This complex, called the death-inducing signaling complex (DISC), causes pro-caspase-8 to be processed and activated. After activating caspase-8, it can process and trigger caspases 3, 6, and 7, which are effector caspases that ultimately cause cell death [67]. Death receptor activation in certain cells causes caspase-8 to cleave the BH3-only protein BID. BID cleavage results in tBID, a 15 kDa shortened active protein that moves to mitochondria and joins the anti-apoptotic BCL-2 protein to form a heterodimer. This connection causes the antiapoptotic BCL-2 proteins to become inactive, which in turn causes the proapoptotic proteins, BAK and BAX, to become active. Activation of BAX and BAK results in the formation of the apoptosome, the activation of caspases, the release of cytochrome c, the permeability of the mitochondrial membrane, and apoptosis.
Granzyme A or B can be used to trigger apoptosis in the perforin/granzyme pathway. Granules in the cytoplasm of NK and CTL cells release serine proteases called granzymes [70, 71]. Furthermore, by making holes in target cells' cell membranes, a glycoprotein known as perforin facilitates granzyme entry [70, 72]. Granzyme B is caspase-dependent in this case, while the Granzyme A (GzmA) pathway induces apoptosis through a caspase-independent mechanism [73]. GzmA plays a critical role in the apoptotic activity of cytotoxic T lymphocytes. Upon entry into the target cell, GzmA cleaves the SET complex, a group of proteins that protects chromatin integrity. This cleavage leads to the release of NM23-H1, a nuclease responsible for promoting DNA degradation during apoptosis. By dismantling the SET complex, GzmA disrupts chromatin structure, thereby facilitating cell death independently of caspase activation. According to these, the inactivation of this complex by granzyme likely encourages apoptosis by impeding the preservation of DNA and chromatin structures [74]. Granzyme B, however, either directly or indirectly activates caspase-3 through caspase-8. Granzyme B cleaves the BID protein specifically, amplifies the death signal, and releases cytochrome c via the mitochondrial pathway [63]. Figure 3 shows these three routes.
3.1. The Role of the BCL-2 Family in Apoptosis
Apoptosis is a strictly controlled type of cell death with unique genetic and biochemical processes that are necessary for healthy tissue development and homeostasis. It helps metazoan organisms maintain the proper balance between cell death and survival by assisting in the removal of unwanted and superfluous cells [58, 74]. Both internal cell damage and signals from the cell surface can cause apoptosis [67]. It has been determined that the BCL-2 family is essential for either triggering or inhibiting the apoptotic process [75].
Transcriptional and/or posttranscriptional activation of BH3-only proteins triggers the BCL-2-regulated apoptotic pathway in response to various upstream signalling events [76]. BH3-only proteins activate BAK and BAX. By competing with pro-survival family members, it can either directly or indirectly activate BAX and BAK [77]. Homodimers and heterodimers produced by activated BAX and BAK interact to create pore-like structures in the outer mitochondrial membrane.
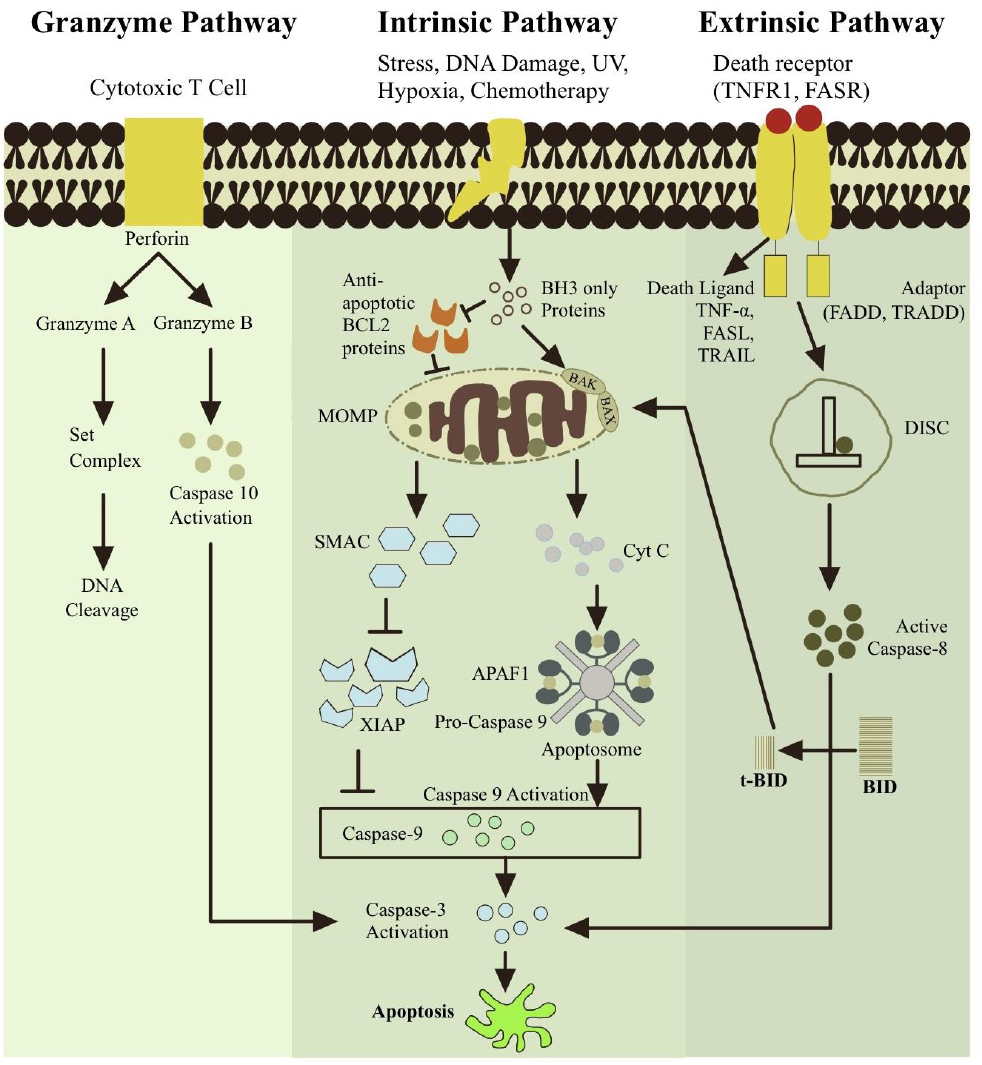
The extrinsic, intrinsic, and perforin/granzyme pathways are the three main classifications of apoptotic pathways. Each pathway is controlled by activating distinct caspases and starts with a distinct signal from the cell that causes death. Death receptors (DRs) and death-inducing ligands interact through their adaptor domains to initiate the extrinsic pathway. This process is then regulated by the multiprotein complex DISC and caspase-8. The stimuli initially activate BH3-only proteins in the intrinsic apoptotic pathway. After stimulating interactions between BCL-2 members resulting in a MOMP complex, cytochrome c is released, triggering the apoptotic signals. Cytochrome c and other caspase activation drive this route. Granzyme A and Granzyme B are secreted by cytotoxic T cells in the perforin/granzyme pathway. The caspase-independent mechanism of the granzyme A pathway. Granzyme B triggers apoptosis by activating caspase-10, which is connected to the intrinsic route. In this case, caspase-3 is eventually activated by all pathways. Nevertheless, apoptosis is ultimately induced by caspase-3 activation. Chromosomes and cytoskeletal proteins are among the vital components of cells that are broken down by caspase-3. Apoptotic bodies form as a result, and phagocytic cells consume and eliminate them before the organism does.
Available online under the terms of the Creative Commons Attribution Non-Commercial License 4.0. [29].
This process, called mitochondrial outer membrane permeabilization (MOMP), enables the release of cytochrome c and other apoptogenic substances into the cytosol, which triggers caspase activation and apoptosis [78].
MOMP (mitochondrial outer membrane permeabilization) not only causes mitochondrial malfunction, leading to depletion of energy reserves, but also results in cell death even when caspases are inactive; thus, recovery from MOMP is possible only under exceptional circumstances, making it a critical event preceding cell death [79]. The induction of apoptosis by BH3-only proteins varies, with some, such as BIM, PUMA, and BID, being more potent inducers than others, such as BAD, NOXA, and BMF [77]. The interactions between proapoptotic BH3-only proteins and prosurvival proteins differ: BIM and PUMA are promiscuous, binding with high affinity to all prosurvival BCL-2 family members, whereas BAD selectively binds to BCL-2, BCL-XL, and BCL-W but not MCL-1 or BFL-1, and NOXA primarily interacts with MCL-1 [80]. This selectivity is also reflected in the design of drugs aimed at promoting apoptosis, which often mimic these specific interactions [76, 80].
3.2. BAX and BAK Activation Models
In healthy cells, BAK is localized to the mitochondria and contains a transmembrane domain (α9) that spans the outer mitochondrial membrane [45]. Conversely, BAX is primarily found in the cytosol, with its transmembrane domain concealed within a hydrophobic groove in its inactive form. Upon encountering cytotoxic signals, BAX translocates to the mitochondria, where it contributes to apoptosis [45]. The mechanisms underlying the activation of BAX and BAK have long been considered a central challenge, often described as the “holy grail” in apoptosis research. During apoptosis, both proteins undergo conformational changes that transform them from harmless precursors into potent effectors capable of permeabilizing the mitochondrial outer membrane [10].
Three hypotheses have been proposed to explain the activation of BAX and BAK: direct activation, indirect activation, and membrane-mediated permissive mechanisms [21, 26]. Some BH3-only proteins are capable of directly activating BAX and BAK; however, while these proteins readily interact with prosurvival BCL-2 family members, detecting early interactions with BAX and BAK has been challenging [81]. In the direct activation model, BH3-only proteins are categorized as either sensitizers or activators. Sensitizer BH3-only proteins bind to and inactivate antiapoptotic BCL-2 proteins, rather than directly activating BAX and BAK [82]. Conversely, activator BH3-only proteins bind directly to BAX and BAK, inducing conformational changes that promote oligomerization and activation of these effectors [21].
According to the indirect activation model, BH3-only proteins neutralize anti-apoptotic proteins, thereby enabling BAK and BAX activation. This process allows apoptosis to be mediated indirectly via BAK and BAX. Under this mechanism, antiapoptotic BCL-2 family members bind to BAK and BAX to inhibit mitochondrial outer membrane permeabilization (MOMP). Instead of directly interacting with BAK and BAX, the primary function of BH3-only proteins is to bind to and displace BAK and BAX from the anti-apoptotic proteins. This mechanism is also referred to as the displacement model [83].
Another hypothesis for BAX and BAK activation is the membrane-mediated permissive paradigm, which involves the mitochondrial outer membrane (MOM). In healthy cells, BAX and BAK typically exist as inactive monomers or dimers. During apoptosis, antiapoptotic BCL-2 family proteins bind to and are neutralized by activated BH3-only proteins on the mitochondrial outer membrane. This neutralization increases the likelihood of spontaneous activation of BAX and BAK, leading to homo-oligomerization and the formation of cytochrome c-releasing pores [84]. Both BAX and BAK are anchored to the membrane via helix 9, which facilitates their spontaneous activation within the lipid bilayer of the MOM. In this context, the primary role of BH3-only proteins is to inactivate anti-apoptotic BCL-2 proteins, aligning with the indirect activation paradigm. However, the membrane-mediated permissive model posits that BH3-only proteins can directly activate BAX and BAK within the membrane [10, 29]. Although BH3-only proteins are considered the main activators of BAX and BAK, other proteins, such as p53, Casp8p41, and retinoblastoma protein, have also been shown to directly activate these effectors. Recent evidence further suggests that active BAX and BAK can, in turn, act as activators of each other [21].
3.3. Formation of Cellular Membrane Pores by BAX and BAK
Cellular membranes serve as essential platforms for storing and transmitting biochemical information required for cell signaling and function. To maintain this functionality, membranes must support controlled exchange between the intracellular and extracellular environment. This is achieved through the presence of specific proteins, such as pore-forming proteins, ion pumps, and cell adhesion molecules, all of which interact with the dynamic properties of the lipid bilayer. Pore-forming proteins (PFPs) are found in all life forms and modify membrane permeability, allowing the passage of ions, small molecules, and occasionally larger biomolecules. In mammals, the BCL-2 family plays a regulatory role in this process during apoptosis and bacterial defense mechanisms [85]. Specifically, the proteins BAX and BAK contribute to mitochondrial outer membrane (MOM) permeabilization during apoptosis, resulting in the release of various mitochondrial proteins into the cytoplasm [86].
In normal physiological conditions, BAX and BAK exist as monomeric proteins with a compact globular structure composed of α-helices. The hydrophobic α5 helix is surrounded by amphipathic helices, while the α9 helix at the C-terminal acts as a transmembrane segment. This α9 domain secures BAK within the MOM under resting conditions. In contrast, BAX remains primarily in the cytosol and only integrates into the MOM after receiving apoptotic stimuli, inserting its α9 region into the membrane. Conformational changes during activation expose the BH3 domain (α2), which later re-engages with activated BAX or BAK molecules. This leads to the formation of symmetric dimers via α6 helix interaction, which then assemble into larger oligomeric complexes [87, 88].
BAX and BAK disrupt the lipid bilayer by forming toroidal pores, where both proteins and lipids contribute to the pore wall. The physical properties of the membrane and the conformational states of BAX and BAK influence the size and stability of these pores [89]. In artificial lipid systems, the pores expand rapidly upon formation and then stabilize at diameters ranging from 3 to 8 nanometers, depending on the concentration and configuration of the proteins. Two categories of BAX-induced pores have been identified: one is voltage-sensitive, small in diameter, selectively permeable to cations, and incapable of protein translocation; the other is voltage-independent and sufficiently large to permit proteins, such as cytochrome c to pass through the membrane [90].
Various structural models explain how BAX and BAK insert into mitochondrial membranes to create toroidal pores [91]. The umbrella model suggests that helices α5 and α6 insert into the lipid bilayer as a hairpin structure that spans the membrane [89, 92]. Another explanation, known as the in-plane model, proposes that α4 and α5 helices lie flat on the membrane surface, causing local membrane curvature and tension, which facilitates pore formation. In this configuration, α9 is fully inserted into the membrane, whereas α5 and α6 are only partially embedded [91].
The clamp model describes BAX oligomers forming dimers in which helices α2 through α5 form a dimerization domain that lines the pore edge, in association with lipid head groups. Since the pore-forming process does not involve full transmembrane insertion of the entire protein, this mechanism may require translocation of BAX monomers to the opposite side of the membrane. In this model, the critical conformational shift involves partial opening of the hairpin formed by the α5 and α6 helices, thereby creating a clamp-like configuration. Helix α5 is directly exposed to the membrane interior, while helix α6 remains at the lipid interface [93]. These structures are essential for mitochondrial outer membrane permeabilization, a key step in apoptotic signaling.
3.4. Apoptotic Pathway Stimulation by BH3-only Proteins
Members of the BCL-2 protein family are defined by the presence of conserved BCL-2 homology (BH) domains, including BH1, BH2, BH3, and BH4. BH3-only proteins, as the name implies, express only the BH3 domain and function either as activators or sensitizers. In contrast, anti-apoptotic and effector proteins contain multiple BH domains [94]. BH3-only proteins function as molecular sensors that detect a variety of stress signals, such as developmental cues or deprivation of growth factors. These stimuli can initiate apoptosis, a tightly regulated process that eliminates damaged or unneeded cells [95].
Eight BH3-only proteins have been extensively investigated in mammalian systems. These include BIM, BAD, BMF, BID, NOXA, BIK, PUMA, and HRK [96]. Their individual functions and overlapping roles have been clarified through gene disruption studies [45]. These proteins either directly activate proapoptotic proteins like BAX or indirectly promote apoptosis by inhibiting anti-apoptotic members of the BCL-2 family. Anti-apoptotic proteins and their viral homologs contain hydrophobic binding grooves formed by their BH domains. BH3-only proteins interact with these sites, and their ability to bind a range of targets is regulated by transcriptional and post-translational mechanisms [97].
BH3-only activators, such as BID, PUMA, and BIM, are capable of directly binding to and activating BAX and BAK. Their activation leads to oligomerization and formation of macropores in the mitochondrial outer membrane. In contrast, BH3-only sensitizers, which include BAD, BIK, NOXA, HRK, and BMF, do not directly activate BAX or BAK. Instead, they bind to antiapoptotic proteins, displacing BH3-only activators or previously activated forms of BAX and BAK, thereby indirectly promoting cell death [98]. The pro-survival activity of BAX and BAK is controlled by six known mammalian anti-apoptotic proteins: BCL-2, BCL-XL, BCL-W, MCL-1, A1, and BCL-B. These proteins are targets of BH3-only inhibition [99].
The primary step in the apoptotic pathway is the oligomerization of BAX and BAK at the mitochondrial outer membrane, leading to membrane permeabilization (MOMP). This allows the release of proapoptotic factors, including cytochrome c, from the mitochondrial intermembrane space, thereby initiating a caspase-dependent proteolytic cascade [65, 99]. The upregulation of BH3-only protein expression frequently triggers MOMP and marks the onset of apoptosis [100].
Among BH3-only proteins, PUMA and NOXA are direct transcriptional targets of p53 and play pivotal roles in apoptosis induced by genotoxic stress, including gamma irradiation and chemotherapeutic drugs [65]. BID must be unfolded and cleaved by caspases to become active. The resulting tBID form acts as a membrane-targeting ligand for BAK and facilitates cytochrome c release. BID, BIM, and PUMA are all capable of directly activating BAX and BAK, while the remaining BH3-only proteins function primarily as derepressors by blocking antiapoptotic interactions [95].
BIM and PUMA have high binding affinities for all members of the anti-apoptotic BCL-2 family, making them effective across a broad range of targets. Other BH3-only proteins exhibit selective binding preferences, sometimes differing by as much as 1000-fold [97]. BH3 mimetics are synthetic molecules developed to mimic BH3-only proteins. These compounds bind with high affinity and specificity to the hydrophobic grooves of anti-apoptotic proteins, blocking their function [94, 98]. ABT-737 was the first BH3 mimetic developed. It binds strongly to BCL-2, BCL-XL, and BCL-W, but shows lower affinity for MCL-1 and A1/BFL1 [101]. ABT-263 (navitoclax) is another potent inhibitor that has shown therapeutic potential in treating chronic lymphocytic leukemia (CLL) [98]. Both ABT-737 and ABT-263 have demonstrated significant proapoptotic effects in experimental models using primary tumor cells [94].
4. THE THERAPEUTIC POTENTIAL OF BCL-2 FAMILY PROTEINS
BCL-2 family proteins are crucial regulators of apoptosis and play a significant role in determining the efficacy of anticancer treatments. Their potential use as biomarkers to predict therapeutic outcomes is currently under exploration [102]. Targeted cancer therapies aim to inhibit the function of specific proteins essential to cancer progression. Among these, anti-apoptotic BCL-2 family proteins are particularly promising due to their elevated expression in malignant cells compared to normal tissues [103]. Notably, the development of a new class of anticancer agents that selectively inhibit these proteins has advanced considerably in recent years [104]. BH3 profiling has emerged as a powerful tool for forecasting treatment responses in various cancers, including the sensitivity of lymphoma cell lines to chemotherapy. This method is particularly effective at identifying tumors with high apoptotic priming, which are more likely to respond positively to both conventional treatments and BH3 mimetics [102]. These mimetics, a novel class of anticancer drugs, imitate the behavior of BH3-only proteins by binding to and neutralizing antiapoptotic BCL-2 family members, thereby triggering apoptosis in cancer cells [76, 105]. When BH3 mimetics bind to the hydrophobic grooves of these anti-apoptotic proteins, they displace bound BH3-only proteins, which in turn activate proapoptotic proteins BAK and BAX. Alternatively, they can also reduce the apoptotic threshold in unprimed cells by occupying these grooves and inhibiting newly produced BH3-only proteins [106]. A range of compounds have been identified as BH3 mimetics, including ABT-737, ABT-263 (navitoclax), ABT-199, GX15-070 (obatoclax), AT-101, gossypol, BI-97D6, TW37, the S1 derivative, BH3-M6, Marinopyrrole A, maritoclax, MIM1, BAM7, A-1155463, and A-1210477 [107].
Among these, ABT-737 stands out as the prototype BH3 mimetic. It was the first molecule intentionally designed to mimic the activity of BH3-only proteins, targeting the antiapoptotic proteins BCL-2, BCL-XL, and BCL-w with high affinity (1 nmol/L). However, it exhibits weaker binding to BCL-B, MCL-1, and A1 [45, 48]. ABT-737 demonstrates broad antitumor activity in multiple cancer types, including multiple myeloma, chronic lymphocytic leukemia (CLL), acute myeloid leukemia, small-cell lung cancer, prostate cancer, and glioblastoma [107].
ABT-263 (navitoclax), a successor to ABT-737, is an orally bioavailable agent that binds BCL-2 family proteins at nanomolar concentrations. It has a stronger binding affinity for BCL-2 and BCL-XL compared to MCL1 and BCL-W [103]. Preclinical studies show its effectiveness when used alongside standard therapies, including efficacy in specific breast cancers [108]. Clinical trials have confirmed its potency as a monotherapy, showing significant tumor reduction in many CLL patients [109]. However, since BCL-XL is crucial for platelet survival, navitoclax use can lead to dose-limiting thrombocytopenia caused by BAX and BAK activation, a concern that also applies to other BCL-XL-selective BH3 mimetics [32]. ABT-199 is the first highly specific inhibitor of BCL-2. It has shown potent anticancer activity in early clinical trials involving CLL and non-Hodgkin lymphoma, offering superior response rates compared to navitoclax and, importantly, does not cause thrombocytopenia [110].
Gossypol, a naturally occurring phenolic compound in cotton plants, has been shown to induce apoptosis in murine tumor models. Its synthetic derivative, AT101, binds to BCL-2, BCL-XL, and MCL-1 [29, 103]. Another derivative of ABT-737, BM-957, also induces apoptosis through caspase-3 activation in cancer cells and promotes tumor regression in xenograft models. It binds to BCL-2 and BCL-XL with sub-nanomolar affinity. BM-1197 functions similarly, triggering BAX/BAK-mediated apoptosis in small-cell lung cancer cell lines and exhibiting improved solubility and pharmacokinetics. It has produced durable, complete tumor regressions in multiple xenograft models [110]. Due to its frequent overexpression in cancers, MCL-1 has become a major target for inhibition. A-1210477 was the first selective MCL-1 inhibitor and showed in vitro efficacy against multiple myeloma [80]. S63845, a more potent and selective agent, has demonstrated substantial single-agent activity in cancers, such as multiple myeloma, lymphoma, chronic myeloid leukemia, and several solid tumors, including non-small-cell lung cancer, breast cancer, and melanoma. Additional promising inhibitors include AMG 176 and AZD5991, both of which have sub-nanomolar binding affinities and exhibit in vivo activity in models of multiple myeloma and acute myeloid leukemia [80].
New anticancer strategies may also emerge from compounds that directly activate BAX or BAK [111]. By targeting their canonical grooves, such agents could promote reversible apoptosis. Compounds that mimic the conformational changes triggered by BH3 peptides can initiate the apoptotic cascade [112]. Recent developments have introduced small molecules that directly bind to and activate proapoptotic BCL-2 family proteins. For instance, BAM-7 directly binds to and activates BAX with high affinity, leading to apoptosis. The BH3-only peptide PUMA activates BAK by binding with nanomolar affinity to its canonical site, causing BAK-dependent mitochondrial outer membrane permeabilization (MOMP) and subsequent cell death. SAHB, a stabilized PUMA-BH3 helix, can inhibit all anti-apoptotic BCL-2 family members and also directly activate BAX [110]. Overall, targeting the BCL-2 family proteins offers vast potential for drug development. The evolution of BH3 mimetics has reached a sophisticated stage, not only enhancing cancer treatment options but also paving the way for therapies in autoimmune diseases [112].
CONCLUSION
This study focuses on the BCL-2 protein family and its role in the regulation of apoptosis. Members of the BCL-2 family either promote or inhibit apoptotic pathways and are categorized into three main groups: pro-apoptotic proteins, anti-apoptotic proteins, and BH3-only proteins. Each category performs distinct biochemical functions. BH3-only proteins primarily act as messengers, transmitting apoptotic signals to other BCL-2 proteins. They activate pro-apoptotic proteins, such as BAX and BAK, which undergo structural changes leading to mitochondrial outer membrane permeabilization (MOMP). Conversely, anti-apoptotic BCL-2 proteins inhibit MOMP and suppress the mitochondrial pathway of apoptosis. Apoptosis is generally triggered through either intrinsic or extrinsic mechanisms, although a third pathway, the perforin/granzyme pathway, has also been identified. These pathways are governed in part by the BCL-2 protein family, underscoring their central role in cell death regulation. Beyond their apoptotic functions, BCL-2 family proteins are also being explored for therapeutic applications, particularly in cancer treatment, where they influence cellular responses to apoptosis-inducing therapies.
Research has demonstrated that BCL-2 family proteins participate in various cellular processes beyond apoptosis, indicating their broader biological relevance. Dysregulation of these proteins has been associated with conditions, such as cancer, neurodegenerative diseases, ischemia, and autoimmune disorders, reaffirming their importance as key regulators of apoptosis [1]. Their involvement in tumor formation, cancer cell survival, and responsiveness to both conventional and targeted therapies highlights their therapeutic potential [102]. The anti-apoptotic functions of BCL-2 proteins are particularly critical in the progression of solid tumors. Consequently, targeting these proteins with specific inhibitors, either as standalone treatments or in combination with standard therapies, presents a promising strategy to overcome resistance in solid tumors and extend remission periods [19]. Additionally, disrupting the interaction between BH3-only proteins and anti-apoptotic BCL-2 members can promote cancer cell death by concurrently neutralizing one or more of these protective proteins [48]. A deeper understanding of the roles of BH3-only proteins in disease has paved the way for new therapeutic approaches, such as the development of BH3 mimetics [95]. These drugs are designed to specifically inhibit anti-apoptotic BCL-2 family proteins. BH3 mimetics hold potential for improving treatments not only for cancer but also for autoimmune and infectious diseases [31].
BCL-2 family proteins are already recognized for their significant biochemical and therapeutic contributions, and they hold substantial promise for future advancements. Targeting this protein family represents a key strategy in the ongoing pursuit of innovative chemical and pharmaceutical developments. BH3 mimetics, in particular, have reached an advanced stage in their development and are being utilized for the treatment of cancers and potentially autoimmune diseases [112]. However, gaps remain in our understanding, especially regarding the diverse isoforms of antiapoptotic BCL-2 proteins. To harness the full therapeutic potential of this pathway, more comprehensive investigations into the various splice variants and their roles in apoptosis are necessary [16].
Additionally, further research is essential to establish the effectiveness of BCL-2 inhibitors in treating both chemotherapy-sensitive and chemotherapy-resistant cancers [113]. Continued efforts are also needed to identify effective drug-like inhibitors specifically targeting BCL-XL and MCL-1, with the goal of enhancing therapeutic outcomes [21]. Looking ahead, successfully targeting anti-apoptotic BCL-2 family members may lead to improved treatment responses and increased survival rates in patients with solid tumors and other malignancies beyond hematologic cancers [19].
AUTHORS’ CONTRIBUTIONS
The authors confirm their contributions to the paper as follows: R.R.: Study conception and design; R.R. and S.A.: Data collection; R.R. and S.A.: Analysis and interpretation of results; Draft manuscript. All authors reviewed the results and approved the final version of the manuscript.
LIST OF ABBREVIATIONS
| BCL-2 | = B-cell lymphoma 2 |
| BFK | = BCL-2 family kin |
| BH | = BCL-2 homology |
| TM | = Transmembrane |
| MOM | = Mitochondrial outer membrane |
| AML | = Acute myeloid leukemia |
| CLL | = Chronic lymphocytic leukemia |
CONFLICT OF INTEREST
Dr. Robin Raufayel is the Editorial Board Member of The Open Medicinal Chemistry Journal.
ACKNOWLEDGEMENTS
Declared none.

