All published articles of this journal are available on ScienceDirect.

Microwave-assisted Green Synthesis and Integrated Bioinformatics Study Reveal Curcumin Analogs Dibenzylidene-cyclohexanones as Novel Potential Anti-Tuberculosis Agents
Authors Info & Affiliations
Abstract
Introduction
Tuberculosis (TB) remains a major global health challenge, further complicated by drug resistance and comorbidities. This study investigates curcumin analogues, dibenzylidene-cyclohexanones, as potential multitarget anti-TB agents.
Methods
Nine benzylidene cyclohexanone derivatives were synthesized using microwave-assisted techniques (yields: 23–81%). Their potential activities were evaluated through molecular docking against key Mycobacterium tuberculosis (Mtb) enzymes and supported by network pharmacology analysis focused on TB-related pathologies.
Results
The benzylidene cyclohexanone analogs A-135 (81%), A-144 (58%), and A-154 (30%), synthesized efficiently via microwave-assisted green chemistry, exhibited superior multitarget binding affinities against key Mtb enzymes—A-135 (MtFabH, pantothenate synthetase), A-144 (MurE, DprE1, PTPs), and A-154 (Ddn, GlmU, Pks13)—with halogen substituents enhancing interactions through halogen bonds and lipophilicity; network pharmacology further revealed 294 overlapping TB-related targets and identified NF-κB1, STAT3, STAT1, and PTGS2 as key hubs mediating their multitarget therapeutic potential modulating TB clinical manifestation in human body.
Discussion
They exhibited superior multitarget binding affinities against key Mtb enzymes, A-135 (MtFabH, pantothenate synthetase), A-144 (MurE, DprE1, PTPs), and A-154 (Ddn, GlmU, Pks13)—with halogen substituents enhancing interactions through halogen bonds and lipophilicity; network pharmacology further revealed 294 overlapping TB-related targets and identified NF-κB1, STAT3, STAT1, and PTGS2 as key hubs mediating their multitarget therapeutic potential, modulating TB clinical manifestation in the human body.
Conclusion
A-135, A-144, and A-154 demonstrate promising multitarget anti-TB activity and potential as adjuvant therapies to complement existing treatments, especially for managing drug resistance and related comorbidities.
1. INTRODUCTION
Tuberculosis (TB), caused by Mycobacterium tuberculosis (Mtb), is transmitted via inhalation of aerosolized droplets and primarily infects alveolar macrophages, where it evades immune responses by inhibiting phagosome-lysosome fusion and neutralizing reactive oxygen species. These mechanisms promote intracellular survival, granuloma formation, and long-term latency, with reactivation often occurring under immunosuppressive conditions, leading to pulmonary cavitation and systemic spread [1-3]. Although initial infection is frequently asymptomatic, active TB typically presents with persistent cough, weight loss, fever, night sweats, and fatigue [3, 4]. Despite global health efforts, TB remains a major cause of mortality, with an estimated 1.25 million deaths in 2023, including 161,000 among HIV co-infected individuals. High-burden countries like India, Indonesia, China, and the Philippines account for over two-thirds of global cases, while diagnostic limitations and rising multidrug-resistant TB (MDR-TB), for which only a small fraction receive effective treatment, continue to undermine disease control efforts [4].
The complex pathology and resistance potential of TB necessitate multifaceted drug regimens targeting heterogeneous bacterial populations. For drug-susceptible TB, the World Health Organization recommends a six-month combination therapy of isoniazid, rifampin, pyrazinamide, and ethambutol, while multidrug-resistant TB (MDR-TB) requires newer, all-oral regimens involving agents, such as bedaquiline, pretomanid, linezolid, and moxifloxacin to enhance adherence, shorten treatment duration, and improve cure rates. However, prolonged use of multiple antibiotics is often associated with adverse effects, such as hepatotoxicity, peripheral neuropathy, and gastrointestinal disturbances, which compromise patient adherence, increase treatment discontinuation, and exacerbate the emergence of resistance [5].
Persistent challenges in TB treatment, including lengthy regimens, adverse drug reactions, and rising multidrug resistance, have spurred interest in natural bioactive compounds as adjunctive therapies. Curcumin, a polyphenol from Curcuma longa, exhibits broad pharmacological properties, such as antioxidant, anti-inflammatory, antimicrobial, and hepatoprotective effects [6-9]. Its immunomodulatory activity, particularly in enhancing T-cell responses, further supports its therapeutic relevance [10]. However, curcumin’s clinical utility has historically been limited by poor bioavailability, prompting the development of nanoparticle-based formulations that significantly enhance its solubility, stability, and systemic absorption [11, 12]. Preclinical studies demonstrate that nano-curcumin can enhance the efficacy of first-line anti-TB drugs, reduce hepatotoxicity, and strengthen host immune responses, indicating its potential to improve adherence, shorten treatment duration, and mitigate relapse and resistance, underscoring its promise as a multifunctional adjuvant in TB therapy [6].
Recent advances in tuberculosis (TB) drug development have increasingly emphasized curcumin analogs— structurally modified derivatives aimed at enhancing chemical stability, biological potency, and target specificity. Among them, monocarbonyl curcumin analogs, in which the native β-diketone moiety is replaced by a monoketone group, have shown improved chemical robustness and bioactivity, exhibiting significant anti-inflammatory, antidiabetic, and antitubercular properties in both in vitro and in vivo studies [13]. Additional structural refinements, including ring-closing modifications and heterocyclic substitutions, have further broadened their therapeutic potential. Concurrently, the strategic halogenation of established anti-TB agents, such as fluorinated derivatives of p-aminosalicylic acid and thioacetazone, has proven effective in enhancing antimycobacterial activity. These halogenated compounds are increasingly incorporated into combination regimens, particularly for combating multidrug-resistant TB strains. Fluorine substitution, in particular, contributes to improved pharmacokinetic profiles by enhancing molecular stability, cellular permeability, and target-binding affinity, thereby increasing overall antimicrobial efficacy [14, 15]. When co-administered with novel therapeutics, such as bedaquiline, delamanid, or pretomanid, these halogenated agents demonstrate synergistic effects, contributing to reduced treatment durations, delayed resistance onset, and improved clinical outcomes in resistant TB cases.
In parallel with pharmacological innovation, there is a growing emphasis on efficient and sustainable synthetic approaches in drug development. While traditional synthetic routes remain widely used, they are often limited by prolonged reaction times, harsh conditions, and suboptimal yields [16]. To overcome these limitations, microwave-assisted synthesis has emerged as a promising green chemistry alternative, utilizing electromagnetic irradiation to generate rapid and uniform heating through dipolar polarization and ionic conduction. This technique significantly accelerates reaction kinetics, offering reaction rate enhancements by several orders of magnitude compared to conventional methodologies. This approach improves reaction efficiency, enhances product purity, reduces side products and waste, and often operates under solvent-free or environmentally friendly conditions. By selectively heating reactants rather than the entire system, microwave synthesis lowers energy consumption and operational risks, establishing itself as a crucial tool for sustainable, reproducible, and safe synthetic workflows [17].
This study explored the therapeutic potential of curcumin analogues as adjuncts in TB treatment using an integrated approach involving chemical synthesis, molecular docking, and network pharmacology analysis. Nine novel analogues (A-125, A-128, A-135, A-137, A-143, A-144, A-150, A-152, A-154) were synthesized and docked against key Mtb protein targets involved in cell wall biosynthesis, energy metabolism, and immune evasion. These nine compounds represent variations in halogen substitution patterns to probe differences in binding affinity and biological relevance. While statistical power calculations are not applicable in this context, the compound number was guided by prior exploratory docking studies of a similar scale in early-stage drug discovery. Proteins, such as MurE, Ddn, GlmU, Pks13, DprE1, MtFabH, PanC, and PtpA were selected based on their critical roles in TB pathogenesis as crucial targets for peptidoglycan biosynthesis and metabolism of Mtb. Lead compounds with strong binding affinities were further analyzed using network pharmacology to predict their interactions with host-pathogen pathways, supporting their potential as immunomodulatory agents for enhancing TB therapy. To ensure the alignment of research methods with the study objectives, an exploratory, non-clinical in silico design was employed, using a quantitative approach centered on synthetic compound evaluation and bioinformatics analysis. The literature supporting target selection and compound validation was obtained through structured searches of PubMed, Scopus, and Web of Science databases using defined keywords and inclusion criteria focused on curcumin derivatives and TB-related targets published between 2015 and 2025. Protein and gene targets were curated from UniProt, GeneCards, SwissTargetPrediction, SuperPred Target Prediction, and NCBI. Interaction networks and pathway enrichment analyses were conducted using the STRING database and Cytoscape software, while the WebGestalt server was used for gene ontology (GO) analysis. This integrative strategy enabled the systematic identification and evaluation of multitarget interactions and host-modulatory potential of the analogues, contributing to the rational design of next-generation anti-TB agents.
2. MATERIALS AND METHODS
2.1. Chemicals, Reagents, and Instrumentation
The series of curcumin analog compounds A-125, A-128, A-135, A-137, A-143, A-144, A-150, A-152, and A-154 were synthesized at the Curcumin Research Center (CRC), Prof. Ritmaleni’s Laboratory, Faculty of Pharmacy, Universitas Gadjah Mada, Indonesia. Microwave-assisted synthesis was performed using a Monowave 400 microwave reactor (Anton Paar, Austria). Aldehydes were obtained from Sigma Aldrich (USA), while cyclohexanone, benzaldehydes, and thin-layer chromatography (TLC) precoated silica gel plates (GF256 type 60) were purchased from Merck (Germany). Several organic solvents, including glacial acetic acid, ethanol, acetone, and water, were procured from Sigma Aldrich (USA). Characterization was conducted using Thin-Layer Chromatography (TLC), Infrared (IR) Spectroscopy, Nuclear Magnetic Resonance (NMR) Spectroscopy, and High-Resolution Mass Spectrometry (HRMS). Detailed synthetic procedures for each compound are provided in the synthesis procedure section and Supplementary Materials.
2.2. Synthesis of Curcumin Analogues via Microwave-Assisted Method
Microwave irradiation utilizes electromagnetic waves to uniformly and rapidly heat reactants, thereby enhancing condensation reactions, such as the formation of benzylidene-cyclohexanone, while reducing energy consumption, reaction time, and the use of hazardous chemicals, in line with green chemistry principles. The synthesis of benzylidene-cyclohexanone compounds, especially benzylidene-cyclohexanone derivatives, typically follows a Claisen-Schmidt condensation strategy. This involves the reaction of substituted benzaldehydes (2 molar equivalents) with cyclohexanone (1 molar equivalent) in the presence of an acid catalyst (HCl), sometimes combined with glacial acetic acid, in a suitable organic solvent. Reactions were carried out in a Monowave 400 microwave synthesizer (Anton Paar), where the reaction mixtures were stirred at 600 rpm and irradiated at 80°C for approximately 9 minutes. The crude products were then isolated by washing with appropriate solvent systems (commonly ethanol-water or acetone-water mixtures) and subsequently purified through recrystallization to afford the final yellow crystalline solids.
For each derivative, substituted benzaldehydes, such as 2-chlorobenzaldehyde, 2-bromobenzaldehyde, 3-bromobenzaldehyde, or 4-chlorobenzaldehyde, were employed, tailoring the reaction to produce specific compounds like 2,6-bis(2’-chlorobenzylidene)cyclohexanone (A-125), 2,6-bis(2’-bromobenzylidene)cyclohexanone (A-128), or 2,6-bis(4’-chlorobenzylidene)cyclohexanone (A-144). After microwave irradiation, products were extracted, washed, and recrystallized using solvent combinations optimized for each compound’s solubility and purity. The final products were characterized using Thin-Layer Chromatography (TLC), melting point determination, Infrared (IR) Spectroscopy, Nuclear Magnetic Resonance (NMR) Spectroscopy, and High-Resolution Mass Spectrometry (HRMS), confirming structural identity and purity (Scheme 1).
2.3. Molecular Docking Studies
Molecular docking analysis was performed using Molecular Operating Environment (MOE) version 2015 (licensed by the Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Universitas Gadjah Mada) to investigate interactions with several essential proteins of Mtb.
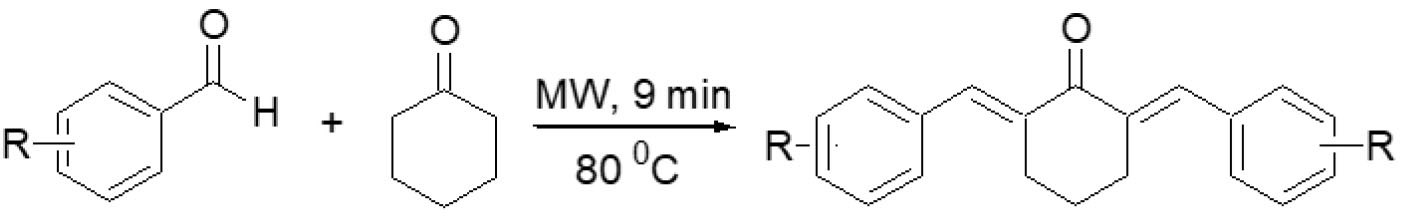
Synthesis of benzylidene-cyclohexanone analogues by microwave irradiation.
The literature supporting docking target selection was obtained through unsystematic exploration of relevant articles across PubMed, Scopus, and Web of Science. Keywords, such as “tuberculosis,” “curcumin analogues,” “molecular docking,” “multitarget therapy,” and “network pharmacology,” were used flexibly to identify studies of interest. Articles were considered if they were published between 2015 and 2025, written in English, and discussed TB-related targets or curcumin-based compounds. Preference was given to peer-reviewed articles containing experimental or computational findings, while reviews without supporting data, case reports, and non-indexed sources were generally excluded. These targets, retrieved from the Protein Data Bank (PDB) via the RCSB website (https://www.rcsb.org), include MurE (PDB ID: 2WTZ), a ligase involved in peptidoglycan synthesis; Ddn (PDB ID: 3R5L), a nitroreductase activating prodrugs; GlmU (4G87), a key enzyme in cell wall precursor biosynthesis; Pks13 (PDB ID: 5V3Y), a polyketide synthase critical for mycolic acid production; DprE1 (PDB ID: 6G83), essential for arabinogalactan synthesis; MtFabH (PDB ID: 2QX1), fatty acid synthase III; Pantothenate Synthetase, involved in coenzyme A biosynthesis; and PTP A (PDB ID: 1U2Q), a virulence factor modulating host immune responses. Protein preparation involved the removal of water molecules, solvents, and cholesterol, followed by the addition of hydrogen atoms using the “Protonate 3D” feature [18-20]. Energy minimization was then performed using the MOE “Energy Minimization” tool with default settings. The docking protocol was validated by locating the binding site with the “Surface and Maps” tool and conducting redocking, with results considered acceptable if the root-mean-square deviation (RMSD) was below 2 Å. The 2D structures of halogenated benzylidene curcumin analogues, along with the reference drugs isoniazid (isonicotinic acid hydrazide/H), pyrazinamide (Z), and ethambutol (E), were converted to SMILES format and subsequently transformed into 3D conformations using the “Builder” function. These compounds were assembled into an MOE database and prepared by assigning partial charges and performing energy minimization. Docking simulations were carried out with the binding site defined as “Ligand Atoms,” placement using the “Triangle Matcher” method, and refinement by “Induced Fit.” The scoring functions applied were “London dG” as the primary scoring parameter and “GBVI/WSA dG” as the secondary. The conformation showing the lowest binding affinity was selected for further analysis and visualized in both 2D and 3D formats.
2.4. Network Pharmacology Analysis
A comprehensive bioinformatics strategy was applied in this study to predict potential protein targets of the combined halogenated compounds A-135, A-144, and A-154, utilizing multiple publicly available target prediction platforms. The network pharmacology approach used in this work was adapted from previous studies, notably the publication “Identification of potential targets of the curcumin analog CCA-1.1 for glioblastoma treatment: integrated computational analysis and in vitro study” [21]. That study successfully integrated network pharmacology with experimental validation, demonstrating the utility of this approach in early-stage drug discovery. Given the favorable correlation between in silico predictions and laboratory findings reported therein, the adopted methodology is considered reliable for preliminary target identification and hypothesis generation in this study. Target prediction and target fishing were performed to map genes associated not only with TB infection but also with its key clinical manifestations, including malnutrition, anemia, inflammatory responses linked to Mtb genes, and multidrug-resistant tuberculosis (MDR-TB). These analyses utilized databases, such as SwissTargetPrediction (http://www.swisstargetprediction.ch), SuperPred Target Prediction (https://prediction.charite.de/subpages/target_ prediction.php), GeneCards (https://www.genecards.org/#), and NCBI (https://www.ncbi.nlm.nih.gov/). Overlapping gene targets were identified through InteractiVenn (https://www.interactivenn.net) to determine potential protein targets involved in TB modulation, referred to as THTGTs (Tuberculosis & Halogenated-Benzylidene Target Genes) [22-27]. Following target prediction, protein-protein interaction (PPI) networks were generated using the STRING database version 11.5 to explore functional associations among the identified proteins. The resulting network data were then analyzed in Cytoscape software version 3.9.1, utilizing the CytoHubba plugin to determine the top 10 hub genes based on the “Degree Score” algorithm [28, 29].
To gain further insights into the biological relevance of these hub genes, functional enrichment analysis was conducted. Gene Ontology (GO) analysis was performed through the WEB-based Gene Set Analysis Toolkit (WebGestalt) (https://webgestalt.org/) using the Over-Representation Analysis (ORA) strategy. The analysis was set to Homo sapiens as the reference organism, and results were filtered by applying a false discovery rate (FDR) threshold (< 0.05) to maintain statistical validity [30].
3. RESULTS AND DISCUSSION
3.1. Chemical Synthesis
Previous studies have shown that halogenated derivatives of anti-TB drugs can offer more effective treatment against Mtb infections, enhancing therapeutic potential compared to non-halogenated compounds. Additionally, these derivatives tend to display lower toxicity, reducing the likelihood of adverse effects experienced by patients. This characteristic makes halogenated derivatives attractive candidates for safer treatment options. Furthermore, halogenated compounds have been found to possess superior pharmacokinetic properties, including improved bioavailability and longer half-lives, which can further improve clinical outcomes.
In the synthetic work conducted, compound A-135 achieved the highest yield, as shown in Fig. (1) and Table 1, reaching 81%, while compound A-152 produced the lowest yield at only 23%. An analysis of the substitution patterns reveals several interesting trends regarding the effect of halogen type and position on yield. Chlorine, when positioned at the ortho site—as seen in entries 1 and 6—tended to generate higher yields compared to its placement at the para position. Moreover, the presence of chlorine in the ortho position (entries 1 and 2) produced a larger yield than bromine positioned similarly, suggesting the positional advantage of chlorine over bromine in this context. However, bromine at the meta position, as observed in entries 2 and 3, gave higher yields than when placed at the ortho position, indicating that bromine benefits from a meta placement. This pattern continued when comparing bromine at the meta versus the para position (entries 3 and 4), where the meta configuration consistently yielded better results. Notably, bromine at the para position performed better compared to chlorine at the same site (entries 4 and 6), while chlorine at the para position outperformed fluorine when both were positioned similarly (entries 6 and 8). Interestingly, when both chlorine and fluorine were present at the ortho position, as seen in entries 5 and 7, the compound containing two chlorine atoms resulted in yields similar to those of compounds containing both halogens. Comparisons between ortho and para substitutions further revealed that chlorine in the ortho position, as in entries 1 and 5, generally produced higher yields than in the para position. However, the reverse was observed in entries 1 and 7, where para-substituted chlorine resulted in better yields. Finally, entries 7 and 9 demonstrated that chlorine in the ortho and para positions yielded comparable results.
Microwave irradiation provides highly efficient energy transfer, promoting faster molecular activation and improved reaction kinetics. This leads to reaction times measured in minutes rather than hours, enhancing laboratory throughput and minimizing solvent waste. Moreover, the uniform heating reduces the formation of byproducts typically associated with local overheating in conventional setups. For benzylidene-cyclohexanone synthesis, this method not only yields good-to-excellent product recovery (23–81% yields) but also enables the scalable preparation of a broad library of derivatives with different aromatic substitutions, facilitating further exploration of their chemical or biological properties.
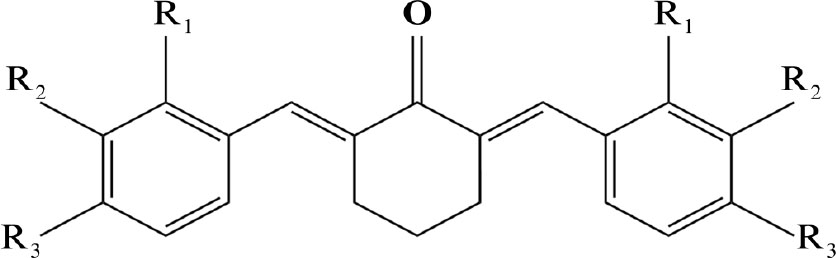
Main backbone chain of halogenated benzylidene cyclohexanone.
| Entry | R-Benzaldehyde | Code | R1 | R2 | R3 | R4 | R5 | Yield (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | 2-Chlorobenzaldehyde | A-125 | Cl | H | H | H | H | 67 |
| 2 | 2-Bromobenzaldehyde | A-128 | Br | H | H | H | H | 56 |
| 3 | 3-Bromobenzaldehyde | A-135 | H | Br | H | H | H | 81 |
| 4 | Bromobenzaldehyde | A-137 | H | H | B | H | H | 74 |
| 5 | 2-Chloro-6-fluorobenzaldehyde | A-143 | Cl | H | H | H | F | 29 |
| 6 | 4-Chlorobenzaldehyde | A-144 | H | H | Cl | H | H | 58 |
| 7 | 2,6-Dichlorobenzaldehyde | A-150 | Cl | H | H | H | Cl | 29 |
| 8 | 4-Fluorobenzaldehyde | A-152 | H | H | F | H | H | 23 |
| 9 | 2,6-Dichlorobenzaldehyde | A-154 | Cl | H | Cl | H | H | 30 |
3.2. Molecular Docking
Molecular docking multitarget assays on Mtb receptors reveal that curcumin analogs, specifically benzylidene cyclohexanone derivatives with halogen substituents, as shown in Fig. (2) and Table 2, demonstrate promising potential as novel anti-TB agents. Among these, compound A-144 exhibited the highest binding affinity, marked by the most negative docking scores toward MurE (-7.21 kJ/mol), DprE1 (-7.6 kJ/mol), and phosphate acetyltransferase (PTP, -5.68 kJ/mol). Compound A-154 emerged as the most promising inhibitor of Ddn (-5.3 kJ/mol), GlmU (-5.31 kJ/mol), and Pks13 (-6.65 kJ/mol), while compound A-128 showed the strongest interactions with MtFabH (-6.96 kJ/mol) and pantothenate synthetase (-5.68 kJ/mol). Notably, these binding affinities surpass those reported for standard anti-TB drugs such as isoniazid, pyrazinamide, and ethambutol, suggesting that a combination of A-135, A-144, and A-154 could offer a potent multitarget approach to TB therapy with superior antimicrobial effects.
Compound A-135 exerts its action by targeting MtFabH and pantothenate synthetase, two enzymes that play vital roles in the maintenance of Mtb’s lipid metabolism and energy production. MtFabH (β-ketoacyl-ACP synthase III) is a key initiator of fatty acid elongation, an essential step in the biosynthesis of mycolic acids, which are critical lipid components of the Mtb cell wall that provide structural rigidity and impermeability to host defenses and antibiotics. Disrupting MtFabH activity cripples the production of these protective lipids, making the bacterium vulnerable to environmental stress. Meanwhile, pantothenate synthetase catalyzes the condensation of pantoate and β-alanine to produce pantothenate (vitamin B5), a precursor to coenzyme A (CoA), which is indispensable for fatty acid synthesis, energy metabolism, and numerous biosynthetic reactions. By inhibiting both MtFabH and pantothenate synthetase, A-128 simultaneously undermines the integrity of the bacterial envelope and deprives the cell of the metabolic fuel required for growth and survival, effectively blocking Mtb’s ability to sustain itself [31, 32].
Compound A-144 acts through inhibition of MurE, DprE1, and PTP, disrupting the construction and maintenance of the Mtb cell wall and its core metabolic processes. MurE is an essential ligase in the peptidoglycan biosynthesis pathway, adding meso-diaminopimelic acid to the growing stem peptide and ensuring the cross-linking that gives bacterial cell walls their mechanical strength. DprE1 is a key epimerase in the synthesis of arabinogalactan, a polysaccharide that bridges the peptidoglycan layer to the mycolic acid-rich outer membrane, a defining feature of the Mtb cell wall. PTPs (protein tyrosine phosphatases) in Mtb are signaling enzymes that regulate phosphorylation-based signaling networks controlling cell division, stress responses, and virulence. Inhibiting PTPs disrupts the bacterium’s ability to coordinate essential cellular processes, impairing its adaptability and survival under hostile conditions. By simultaneously impairing these three targets, A-144 dismantles the bacterium’s ability to synthesize a stable cell wall and generates a severe metabolic bottleneck, causing failure of essential biosynthetic and energetic pathways, ultimately leading to bacterial death [33-35].
| Mycobacterium Tuberculosis Targets | A-125 | A-128 | A-135 | A-137 | A-143 | A-144 | A-150 | A-152 | A-154 | H | Z | E | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mur E (2WTZ) | Dock. Score | -6.86 | -6.69 | -6.12 | -6.84 | -6.78 | -7.21 | -6.18 | -6.57 | -6.43 | -4.79 | -4.64 | -5.77 |
| Residues | Ser 155, Gly 156, Lys 157, Thr 159, Glu 220, Arg 230, Arg 377, Asp 392, Tyr 393, Lys 396 | ||||||||||||
| Ddn (3R5L) |
Dock. Score | -4.53 | -4.80 | -5.04 | -5.13 | -4.50 | -4.86 | -4.63 | -4.68 | -5.30 | -3.91 | -4.92 | -4.00 |
| Residues | Tyr 65, Ser 78, Lys 79, Gly 89, Ser 132, Asp 135 | ||||||||||||
| GlmU (4G87) | Dock. Score | -5.13 | -4.61 | -4.96 | -5.00 | -4.71 | -4.79 | -4.31 | -4.38 | -5.31 | -6.33 | -7.04 | -5.29 |
| Residues | Arg 19, Thr 168, Gln 205, Glu 207 | ||||||||||||
| pks 13 (5V3Y) | Dock. Score | -5.6 | -5.55 | -5.54 | -5.42 | -5.91 | -5.7 | -5.92 | -5.73 | -6.65 | -4.23 | -4.48 | -4.17 |
| Residues | Ile 1507, Tyr 1582, Val 1614, Gln 1633, Thr 1645 | ||||||||||||
| DprE1 (6g83) | Dock. Score | -7.55 | -7.37 | -7.54 | -7.3 | -6.8 | -7.6 | -6.55 | -7.52 | -7.51 | -5.05 | -6.59 | -4.5 |
| Residues | Leu 56, Tyr 60, Thr 122, Gly 182, Ile 184 | ||||||||||||
| MtFabH (2qx1) | Dock. Score | -6.17 | -6.64 | -6.96 | -6.75 | -6.1 | -6.6 | -6.84 | -6.66 | -6.76 | -4.22 | -5.12 | -4.59 |
| Residues | Asp 27, Trp 32, Arg 151, Gly 152, Gly 209, Asn 274, Tyr 304 | ||||||||||||
| Phantotenate Synthethase | Dock. Score | -7.38 | -7.01 | -8.17 | -7.23 | -6.6 | -7.66 | -4.91 | -7.68 | -6.75 | -4.81 | -5.63 | -4.08 |
| Residues | Pro 38, His 44, Phe 156, Gly 158, Gln 164, Met 195 | ||||||||||||
| PTP (1u2q) | Dock. Score | -5.47 | -5.41 | -5.46 | -5.48 | -5.39 | -5.68 | -5.53 | -5.36 | -5.56 | -4.39 | -5.61 | -4.51 |
| Residues | Thr 12, Ile 15, Arg 17, Trp 48, Asn 92 | ||||||||||||
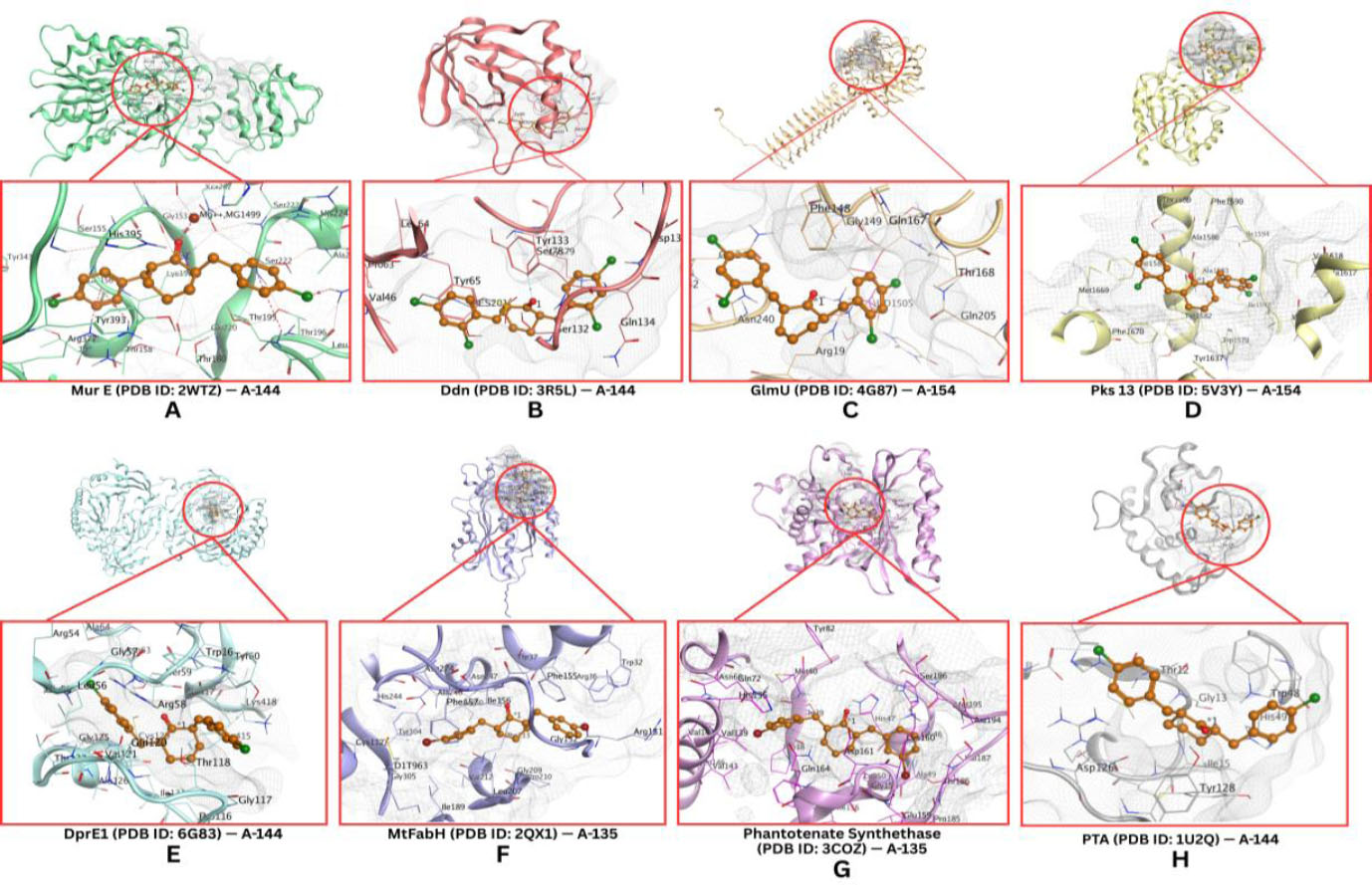
The molecular docking results of halogenated benzylidene-cyclohexanone compounds against Mycobacterium tuberculosis (Mtb) receptors showed the highest affinities for compound A-135 with MtFabH and pantothenate synthetase; compound A-144 with MurE, DprE1, and PTP; and compound A-155 with Ddn, GlmU, and Pks1.
Compound A-154, the most promising inhibitor for Ddn, GlmU, and Pks13, acts on multiple fronts essential to Mtb’s survival under stress and its envelope biosynthesis. Ddn (deazaflavin-dependent nitroreductase) is crucial for activating nitroimidazole prodrugs and managing nitrosative stress, a defense mechanism Mtb relies on to withstand the hostile environment within host macrophages. Inhibiting Ddn compromises the bacterium’s resilience against oxidative and nitrosative stress, rendering it more susceptible to immune clearance. GlmU is a bifunctional enzyme catalyzing the final steps of UDP-N-acetylglucosamine synthesis, a fundamental building block for peptidoglycan assembly. Pks13, on the other hand, catalyzes the final condensation of fatty acyl chains to produce mycolic acids, pivotal components of the mycobacterial outer membrane. By targeting Ddn, GlmU, and Pks13, A-154 simultaneously impairs the bacterium’s stress response system, blocks the supply of cell wall precursors, and halts mycolic acid biosynthesis, striking at the heart of Mtb’s structural integrity and survival machinery [36-38].
The chlorine (Cl) and bromine (Br) substituents on these compounds play an important role in enhancing their biological activity. The halogen atoms increase the molecules’ lipophilicity, facilitating better membrane permeability and stronger interactions within the hydrophobic pockets of their target enzymes. The larger atomic radius and polarizability of bromine in A-135 allow it to establish more potent van der Waals and hydrophobic interactions, whereas the chlorine atoms in A-144 and A-154 contribute electron-withdrawing effects that stabilize halogen bonding and strengthen electrostatic and dipole interactions at the binding sites. Additionally, the presence of two chlorine substituents in A-154 may confer enhanced spatial fitting within the enzyme active sites, increasing its affinity and specificity. Collectively, the incorporation of these halogens fine-tunes the compounds’ binding profiles and improves their overall potency as multitarget anti-TB agents [39, 40]. Together, the superior binding affinities observed for these curcumin analogs, compared to frontline TB drugs (HZE), highlight their potential to serve as a new generation of multitarget anti-TB agents. The combination of A-128, A-144, and A-154, by collectively addressing multiple indispensable enzymatic systems, holds promise for enhanced antimicrobial potency and a reduced likelihood of resistance development. The benzylidene cyclohexanone compounds exhibiting the highest affinity toward Mtb targets were those synthesized in high yields via the microwave-assisted, green chemistry-based approach, namely A-135 (81%) and A-144 (58%). Although A-154 was obtained in a relatively lower yield of 30% compared to these two, this result still underscores the effectiveness of the microwave-assisted synthesis method in the context of novel drug discovery, particularly for potential anti-TB agents, while simultaneously offering an environmentally friendly synthetic route. Although other analogs were not highlighted in detail, they nonetheless demonstrated promising activity, with binding affinities generally surpassing those of frontline anti-Mtb drugs (HZE), thus warranting further investigation and development.
3.3. Network Pharmacology
This study employed a comprehensive bioinformatics strategy utilizing network pharmacology to elucidate the complex interactions between genes implicated in TB and the therapeutic potential of halogenated benzylidene curcumin analogues A-135, A-144, and A-154. Through this approach, novel therapeutic targets, relevant biological pathways, and putative hub genes were identified, particularly those involved in key clinical manifestations of TB, thereby offering valuable insights for future therapeutic development. Among these manifestations, malnutrition, anemia, and inflammatory responses were emphasized due to their critical roles in disease progression, treatment response, and clinical prognosis. Malnutrition compromises host immunity and increases susceptibility to active TB by impairing immune defense mechanisms [41]. Anemia is commonly reported in TB patients and is linked to disease severity, potentially driven by chronic inflammation and disrupted iron homeostasis via the hepcidin pathway [42, 43]. Furthermore, inflammatory responses triggered by Mtb can lead to extensive tissue damage and persistent immune activation, exacerbating disease pathology [44]. The inclusion of multidrug-resistant TB (MDR-TB) as a focal concern is justified by its increasing prevalence and the clinical challenges it presents, including limited treatment options and higher mortality rates [45].
Considering the clinical relevance of these manifestations, they were incorporated into the network pharmacology analysis as primary reference points for target mapping and pathway enrichment. Target gene prediction was performed using SwissTargetPrediction, SuperPred, GeneCards, and NCBI databases to identify genes associated not only with TB itself but also with its associated comorbidities. This approach enabled the identification of overlapping genes modulated by the halogenated compounds, reinforcing their potential utility in modulating multifaceted aspects of TB pathology.
Based on predictions from SwissTargetPrediction, the combination of A-135, A-144, and A-154 was associated with 300 potential molecular targets (Supplementary Table 1). In parallel, the SuperPred Target Prediction platform identified 370 receptor-level targets for the same compound set (Supplementary Tables 2 and 3). GeneCards and NCBI further contributed a comprehensive list of 19,587 genes associated with key clinical manifestations of TBB, including malnutrition, anemia, inflammatory responses triggered by Mtb, and multidrug-resistant TB (MDR-TB) (Supplementary Table 4). Integrative analysis using a Venn diagram revealed 294 overlapping genes, referred to as Tuberculosis and Halogenated-Benzylidene Target Genes (THTGTs), which were selected for subsequent downstream functional and network analyses (Fig. 3A-3D).
The PPI network illustrating the interconnections among the proteins is shown in Fig. (3C) (Supplementary Tables 5 and 6). The hub gene of this compound compilated was identified, with the top 10 genes based on degree score: EGFR, HSP90AA1, STAT3, PTGS2, HSP90AB1, NF-κB1, EP300, APP, STA1, and NR3C1 (Fig. 3D). Bioinformatic analysis shows that combination of these three compounds holds significant potential in overcoming TB and its clinical manifestation through 4 principal proteins identified on high degree score: NF-κB1, STAT3, STAT1, and PTGS2. These proteins play a significant role in regulation and their relevance to TB-associated clinical manifestations (Fig. 4).
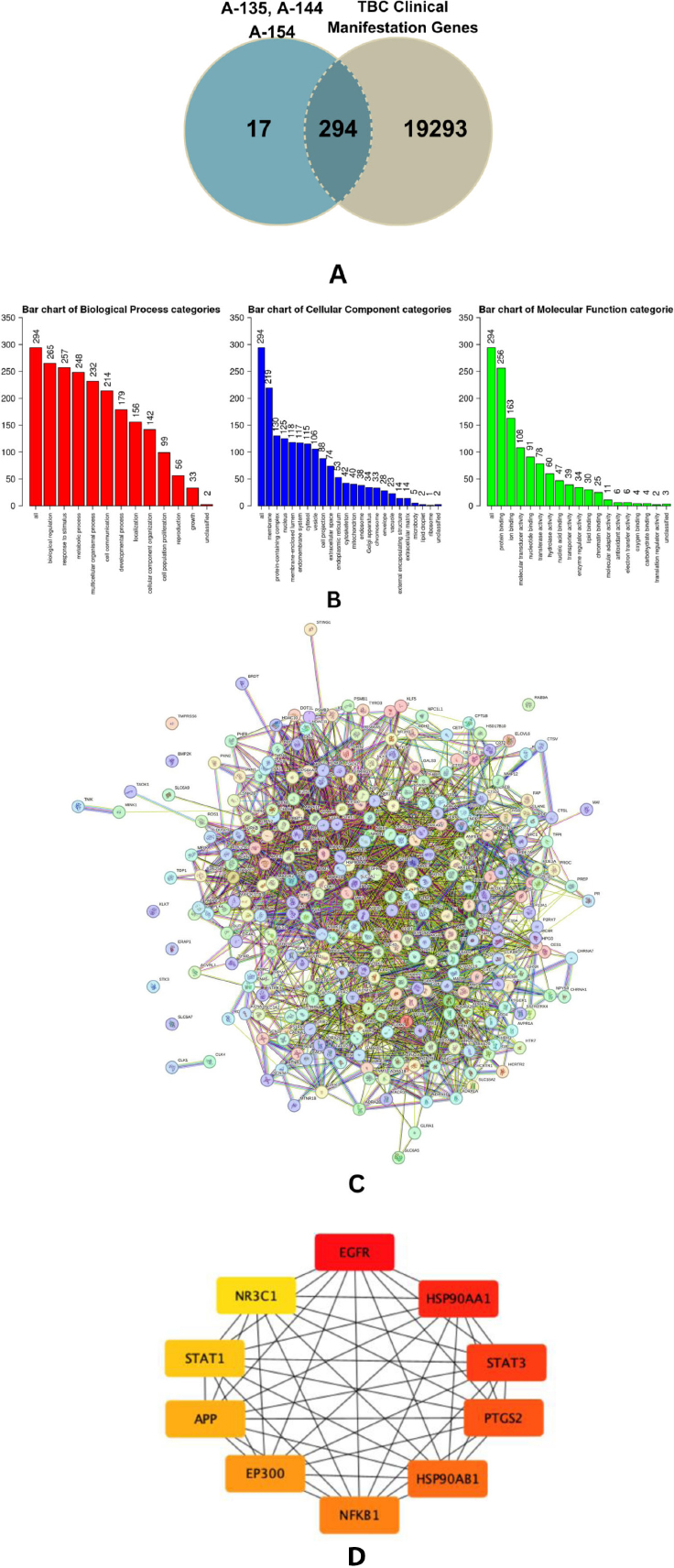
(A) Venn diagram showing 294 predicted target genes identified as THTGTs. (B) GO enrichment analysis of PTPMs categorized into Biological Process (BP), Cellular Component (CC), and Molecular Function (MF). (C) Protein–protein interaction (PPI) network of THTGTs constructed using STRING, consisting of 293 nodes and 2,324 edges. The average local clustering coefficient is 0.405, indicating a high level of interconnectivity among the targets. (D) Top 10 hub genes ranked by degree score, as determined using the CytoHubba plugin in Cytoscape. Node color reflects the degree score, with red indicating the highest, followed by orange and yellow.
To better elucidate the potential mechanism of action and therapeutic relevance of the curcumin analogues identified in this study (A-135, A-144, and A-154), Gene Ontology (GO) enrichment analysis was performed across the domains of Biological Process (BP), Molecular Function (MF), and Cellular Component (CC). GO analysis serves as a critical bioinformatic tool to interpret the functional landscape of target genes and their roles in disease-relevant pathways, particularly in host-pathogen interactions, such as in TB (Fig. 3B) [46, 47]
Gene Ontology (GO) enrichment analysis was conducted to characterize the functional landscape of predicted target genes modulated by halogenated curcumin analogs A-135, A-144, and A-154. In WebGestalt, GO visualization is typically presented as bar plots where the x-axis denotes enriched GO terms, ranked by statistical significance, and the y-axis represents the number of genes associated with each term. A higher gene count indicates a stronger degree of enrichment and reflects greater biological relevance of the term within the dataset [30].
Under the Biological Process (BP) category, the most significantly enriched terms included biological regulation (265 genes), response to stimulus (257 genes), and metabolic processes (248 genes). The high number of genes involved in biological regulation suggests that these compounds may influence core regulatory networks, including transcriptional control, cytokine signaling, and homeostatic adaptation—processes frequently hijacked during Mtb infection. Similarly, enrichment in response to stimulus indicates potential roles in modulating host defense mechanisms and cellular stress pathways, while the metabolic process enrichment suggests involvement in altering bioenergetic and metabolic reprogramming, a key survival strategy of intracellular pathogens. Within the Molecular Function (MF) domain, top-enriched terms included protein binding (256 genes), ion binding (163 genes), and molecular transducer activity (108 genes). pattern implies that the target compounds may modulate protein–protein interactions and receptor-mediated signaling, consistent with their predicted engagement with immune-regulatory proteins, such as STAT1 and PTGS2 (COX-2). These molecular functions are essential for transmitting extracellular signals to intracellular responses during infection and inflammation. The Cellular Component (CC) analysis revealed that gene products were predominantly localized to the membrane (219 genes), protein-containing complexes (130 genes), and the nucleus (125 genes). This localization profile supports the multi-compartmental mechanisms of action, including membrane-bound receptor activation (e.g., toll-like receptors), intracellular complex formation (e.g., inflammasomes), and nuclear transcriptional regulation. The presence of PTGS2 and STAT1 in these compartments further reinforces the mechanistic plausibility of their roles as key molecular targets. Collectively, these GO enrichment results underscore the dual antimicrobial and immunomodulatory potential of the curcumin analogs in host–pathogen interactions [48, 49].
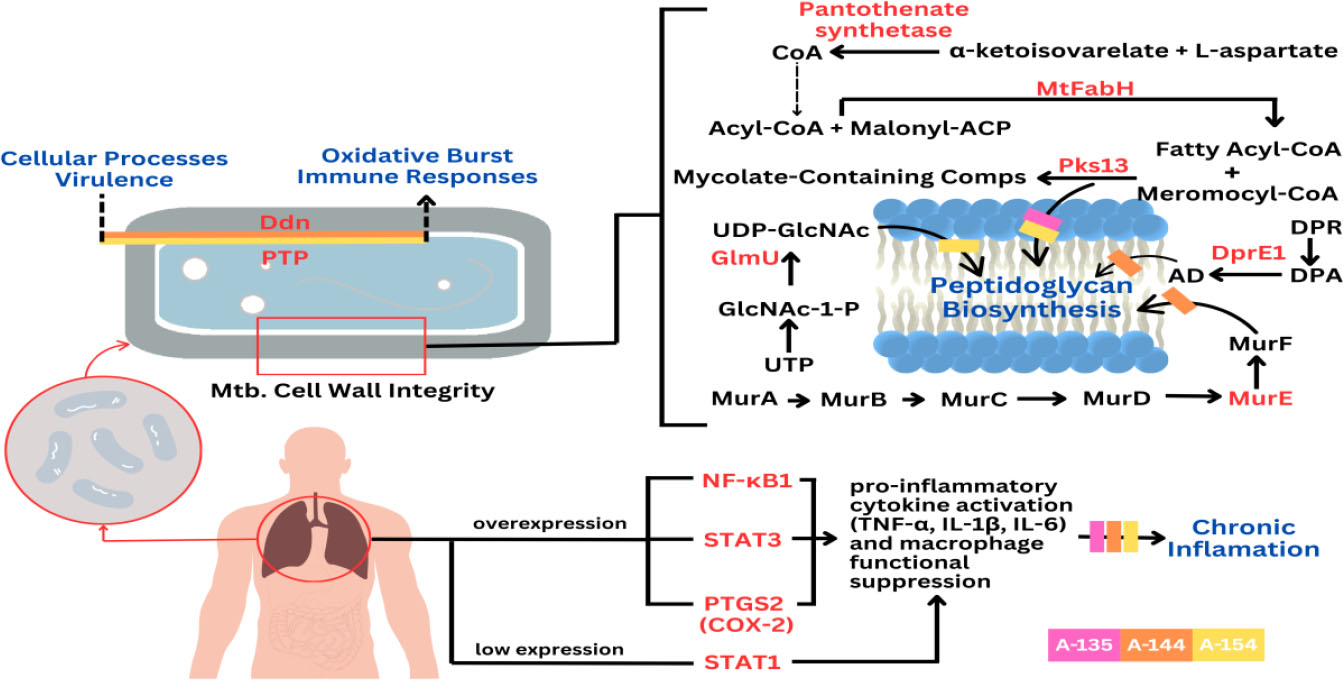
Proposed mechanism of action of combined halogenated benzylidene-cyclohexanone analogs a-135, a-144, and a-154 in counteracting Mycobacterium tuberculosis (Mtb) survivability and the chronic inflammatory manifestations of tuberculosis.
Nuclear Factor Kappa B Subunit 1 (NF-κB1), a key transcription factor in the NF-κB family, plays a pivotal role in host defense and the pathogenesis of TB. During Mtb infection, alveolar macrophages detect pathogen-associated molecular patterns (PAMPs) and secrete pro-inflammatory cytokines, particularly TNF-α. The binding of TNF-α to TNFR1 triggers a signaling cascade involving IκB kinase (IKK), which phosphorylates and degrades IκB, enabling NF-κB translocation into the nucleus. This activates transcription of genes encoding pro-inflammatory cytokines (e.g., IL-1β, IL-6, IL-12, TNF-α), chemokines, and macrophage-activating molecules. NF-κB-driven inflammation is essential for granuloma formation, a hallmark of TB that restricts Mtb spread, while also promoting M1 macrophage polarization, Th1/Th17 responses, and inflammasome activation. However, excessive NF-κB activation may lead to pathological inflammation and clinical symptoms of active TB, whereas insufficient signaling compromises granuloma integrity and bacterial control [50, 51]. In this context, curcumin analogues A-135, A-144, and A-154 tested in this study exhibit inhibitory effects on NF-κB1, potentially by targeting upstream events, such as IKK activation or IκB degradation. These findings support their potential as adjuvant candidates for TB therapy by modulating NF-κB-mediated immunopathology without replacing standard antimicrobial treatment.
Signal Transducer and Activator of Transcription 3 (STAT3) is a central transcription factor that plays a crucial role in modulating immune responses during Mtb infection. STAT3 is activated by cytokines, such as IL-10 and IL-6, secreted by immune cells during Mtb infection, and upon activation, it translocates to the nucleus where it regulates genes involved in cell proliferation, differentiation, and immune regulation. In tuberculosis, STAT3 activation occurs in both infected macrophages and bystander immune cells, exerting complex immunomodulatory effects. Specifically, activated STAT3 suppresses the expression of key pro-inflammatory cytokines, including IL-6, TNF-α, IFN-γ, and MIP-1β, and inhibits nitric oxide (NO) production by repressing inducible nitric oxide synthase (iNOS). This suppression limits the microbicidal activity of macrophages, facilitating intracellular survival of Mtb [52]. In this study, based on in silico analyses, we evaluated a combination of compounds, A-135, A-144, and A-154, that may inhibit STAT3 activation by interfering with its phosphorylation and nuclear translocation, potentially restoring pro-inflammatory mediator expression and enhancing macrophage antimicrobial functions. These findings suggest that targeting STAT3 with this compound combination could serve as a promising adjuvant therapeutic strategy in tuberculosis, pending further experimental validation.
Signal Transducer and Activator of Transcription 1 (STAT1) plays a central role in host defense against Mtb by mediating interferon-gamma (IFN-γ) signaling. Upon IFN-γ stimulation, STAT1 undergoes phosphorylation, dimerization, and nuclear translocation to activate interferon-stimulated genes (ISGs) via gamma-activated sequence (GAS) elements. This cascade induces macrophage activation, nitric oxide production, and apoptosis in infected cells, thereby limiting Mtb replication. STAT1 also promotes M1 macrophage polarization, enhancing bactericidal activity. In early TB infection, phosphorylated STAT1 drives pro-apoptotic signaling, whereas later stages see an accumulation of unphosphorylated STAT1, which disrupts JAK1 phosphorylation and impairs apoptosis, facilitating immune evasion. STAT1 deficiency is linked to increased TB susceptibility, impaired granuloma formation, and higher bacterial loads.
Additionally, STAT1-related molecules and regulatory RNAs have emerged as potential biomarkers for active TB. Despite its importance, Mtb can subvert STAT1 responses by modulating transcriptional coactivators, such as CBP/p300 and selectively suppressing IFN-γ-responsive genes [53, 54]. Interestingly, the combination of curcumin analogues A-135, A-144, and A-154 tested in this study demonstrates a potential to enhance STAT1 activity, possibly by promoting its phosphorylation and nuclear translocation, thereby counteracting Mtb's immune evasion strategies. While the precise molecular mechanism remains to be fully elucidated, this combination may act by facilitating upstream kinase signaling or stabilizing STAT1–DNA binding. These findings suggest a possible role for these compounds as adjunctive therapeutic candidates, aimed at reinforcing host immune responses in conjunction with conventional anti-TB drugs.
Prostaglandin-endoperoxide synthase 2, commonly known as cyclooxygenase-2 (COX-2), is a key enzyme in the biosynthesis of prostanoids, notably prostaglandin E2 (PGE2), and plays a significant role in modulating the immune response during Mtb infection. In tuberculosis, COX-2 is upregulated in monocytes and macrophages upon infection, leading to increased production of PGE2, which acts as a potent immunomodulator [55, 56]. The COX-2/PGE2 axis has complex and context-dependent effects. In the early stages of infection, PGE2 may help limit excessive inflammation and promote macrophage apoptosis, which can be protective by preventing excessive tissue damage and limiting bacterial spread. However, in chronic or advanced TB, elevated PGE2 can suppress Th1 effector responses, dampen macrophage activation, and promote the expansion of regulatory T cells (Tregs), which may impair the host’s ability to control Mtb and contribute to disease progression. Studies have shown that inhibition of COX-2 with drugs, such as indomethacin or etoricoxib, can reduce the number of Mtb-specific Tregs and modulate cytokine production, but may also impair the macrophage’s ability to control mycobacterial infection, suggesting that COX-2 activity is essential for optimal immune function in certain contexts. Clinically, dysregulated COX-2/PGE2 signaling is associated with both protective and detrimental outcomes. At the same time, it can limit harmful inflammation and tissue destruction; excessive or prolonged activation may contribute to immune evasion by Mtb, increased bacterial burden, and more severe disease manifestations such as persistent fever, weight loss, and impaired granuloma formation [55-57]. Interestingly, our findings suggest that curcumin analogues A-135, A-144, and A-154 may attenuate PTGS2 expression, possibly by interfering with upstream NF-κB and MAPK signaling pathways, thereby reducing PGE2 production and restoring macrophage immunocompetence. Although the precise mechanism remains to be fully elucidated, this potential mode of action highlights the promise of these compounds as adjunctive agents in host-directed TB therapy.
| Rank | Gene Symbol | Gene Name | Degree Score |
|---|---|---|---|
| 1 | EGFR | Epidermal Growth Factor Receptor | 90 |
| 2 | HSP90AA1 | Heat Shock Protein 90 Alpha Family Class A Member 1 | 86 |
| 3 | STAT3 | Signal Transducer and Activator of Transcription 3 | 84 |
| 4 | PTGS2 | Prostaglandin-Endoperoxide Synthase 2 | 71 |
| 5 | HSP90AB1 | Heat Shock Protein 90 Alpha Family Class B Member 1 | 69 |
| 6 | NF-κB1 | Nuclear Factor Kappa B Subunit 1 | 65 |
| 7 | EP300 | E1A Binding Protein P300 | 58 |
| 8 | APP | Amyloid Beta Precursor Protein | 51 |
| 9 | STAT1 | Signal Transducer And Activator Of Transcription 1 | 50 |
| 10 | NR3C1 | Nuclear Receptor Subfamily 3 Group C Member 1 | 45 |
In this study, we successfully synthesized novel curcumin analogues using a microwave-assisted method aligned with green chemistry principles, and systematically profiled their potential anti-TB properties through an integrative in silico approach. By targeting Mtb proteins via molecular docking and evaluating interactions with host-related targets through network pharmacology, we identified three promising candidates — A-135, A-144, and A-154 —that exhibit favorable binding profiles and multitarget interactions relevant to TB pathogenesis and clinical manifestations. These compounds hold potential as adjunctive anti-TB agents, yet further validation through in vitro and in vivo studies is essential to confirm their biological efficacy and safety. Moreover, given the inherent formulation challenges commonly associated with curcumin and its derivatives, future efforts should also prioritize optimizing delivery systems to enhance bioavailability and therapeutic performance (Table 3).
CONCLUSION
This study demonstrates the efficient microwave-assisted synthesis of halogenated benzylidene cyclohexanone curcumin analogs, notably A-135 (yield: 81%), A-144 (yield: 58%), and A-154 (yield: 30%), which exhibited strong binding affinities against key Mycobacterium tuberculosis enzymes and disrupted critical pathways, such as cell wall biosynthesis and lipid metabolism. Network pharmacology analysis further indicated their potential to modulate host immune responses via hub proteins NF-κB1, STAT1, STAT3, and PTGS2, supporting a dual antimicrobial and host-directed mechanism. While promising, this study is limited by its in silico nature, which, while powerful for early-stage screening, lacks experimental validation in biological systems. The selection of targets relied on available databases and known Mtb enzymes, which may not capture emerging or less-characterized pathways. The network pharmacology analysis, while comprehensive, is based on predicted interactions, which may not reflect true in vivo dynamics. Additionally, while nine analogs were synthesized, only three were discussed in detail, leaving potential unexplored insights into the structure-activity relationship (SAR) of the remaining compounds. Future work should include experimental assays for target validation in cellular and animal models.
AUTHORS’ CONTRIBUTIONS
The authors confirm their contributions to the paper as follows: R.R.: Study conception and design; I.R., M.R.I., M.T.A.: Data collection; H.G.: Validation; P.M.: Analysis and interpretation of results; R.D.P.A.: Draft manuscript. All authors reviewed the results and approved the final version of the manuscript.
LIST OF ABBREVIATIONS
| TB | = Tuberculosis |
| MDR-TB | = multidrug-resistant TB |
| GO | = Gene Ontology |
| CRC | = Curcumin Research Center |
| TLC | = thin-layer chromatography |
| IR | = Infrared |
| NMR | = Nuclear Magnetic Resonance |
| HRMS | = High-Resolution Mass Spectrometry |
AVAILABILITY OF DATA AND MATERIALS
All data generated or analyzed during this study are included in this published article.
FUNDING
This work was supported by a grant from the Program of Post-Doctoral Batch II Research Grant 2022, Gadjah Mada University, Indonesia, Grant Number: 13602/UN1.P.II/Dit-Lit/PT.01.04/2022, 2 December 2022
ACKNOWLEDGEMENTS
Declared none.

