All published articles of this journal are available on ScienceDirect.

QSAR and Docking Based Screeningof Pyrrolidine Derivatives as Matrix Metalloproteinase-2 Inhibitors
Abstract
Introduction
Cancer remains a leading global health challenge, with drug resistance, toxicity, and economic burden limiting the effectiveness of existing therapies. Matrix metalloproteinase-2 (MMP-2), a key gelatinase involved in extracellular matrix (ECM) degradation, plays a crucial role in cancer metastasis and represents a promising target for anticancer drug development.
Materials and Methods
This study focuses on designing novel MMP-2 inhibitors by employing a comprehensive 2D-Quantitative Structure-Activity Relationship (2D-QSAR) analysis of 71 pyrrolidine derivatives with reported anticancer activity. Docking studies using Autodock Vina software were performed, followed by ADMET analysis using the SwissADME server.
Results
A robust QSAR model was developed using multiple linear regression (MLR) analysis, demonstrating high reliability, statistical significance, and predictive accuracy (r = 0.918, r2cv = 0.842, r2pred= 0.798).
Discussion
Based on QSAR insights, new pyrrolidine derivatives were designed, and their anticancer potential was evaluated through molecular docking studies against MMP-2 (PDB ID: 1HOV). ADMET analysis revealed favorable pharmacokinetic and toxicity profiles for all of the designed compounds. Docking studies showed strong binding affinities, highlighting the potential of these compounds as selective and potent MMP-2 inhibitors.
Conclusion
An integrative approach using QSAR modeling, molecular docking, and ADMET analysis provides a valuable framework for designing effective anticancer agents targeting MMP-2.
1. INTRODUCTION
Matrix metalloproteinases (MMPs) are a large family of calcium-dependent zinc-containing endopeptidases, which are responsible for the tissue remodeling and degradation of the extracellular matrix (ECM), including collagens, elastins, gelatin, matrix glycoproteins, and proteoglycan [1]. Overactivation of MMPs results in an imbalance between the activity of MMPs and tissue inhibitors of metalloproteinases (TIMPs) that can lead to a variety of pathological disorders [2, 3]. There are now 27 MMPs identified. Based on their substrate specificity, these have been divided into subfamilies. Their overexpression is associated with several diseases: cancer, cardiovascular diseases, osteoarthritis, rheumatoid arthritis, chronic obstructive pulmonary disease, psoriasis, dermatitis, Alzheimer´s disease, and periodontitis, among others [1, 4].
In recent years, cancer has become the main cause of death and it is a serious threat to human health, so the development of new, selective and safe anticancer drugs is still the focus of medical research [5]. Degradation of extracellular matrix (ECM) is crucial for malignant tumour growth, invasion, metastasis and angiogenesis [6]. The involvement of MMPs in ECM degradation has been clearly demonstrated. MMPs constitute a family of structurally related zinc-containing endopeptidases that catalyze the hydrolysis of the peptide bond [7]. Gelatinase-A is an important member of this family known as MMP-2 [8]. MMP-2 is massively upregulated in malignant tissues [9]. This is expressed during carcinogenesis and angiogenesis, and it has become clear that this is involved at almost all stages of tumour progression from initial tumour development, growth, and angiogenesis to invasion, metastasis, and growth at secondary sites [10]. Among the MMPs, MMP-2 has been considered a promising target for cancer therapy on the basis of its massive up-regulation in malignant tissues and its unique ability to degrade ECM components [11]. Therefore, the design and development of MMP-2 inhibitors have attracted medicinal chemists; consequently, their synthetic inhibitors were rapidly developed and routed into human clinical trials. But the result has not been very encouraging [12, 13]. Some of the reported MMP inhibitors were introduced into clinical trials, and many of them have shown a good response in cancer treatment. Some promising inhibitors include prinomastat (AG3340), marimastat (BB-2516), S-3304, and cipemastat (Ro32-3555) (Fig. 1) [14].
Towards the design of potent MMP-2 inhibitors, we have made a quantitative structure-activity relationship (QSAR) study on a series of MMP-2 inhibitors. The QSAR studies provide the rationale for drug design by indicating the physicochemical and structural properties of the molecules that control their activities and providing insight into the nature of drug-receptor interactions. QSAR describes the varying role of molecular descriptors on the biological activity of a set of molecules. A model containing those calculated descriptors can be used to predict responses from new compounds, constituting an important tool to support the synthesis of new drugs [15-17]. Thus, considering the continuous need for new anticancer drugs, a QSAR study based on 71 pyrrolidine derivatives (Table 1) synthesized and assayed by Cheng et al. was carried out [18-20].
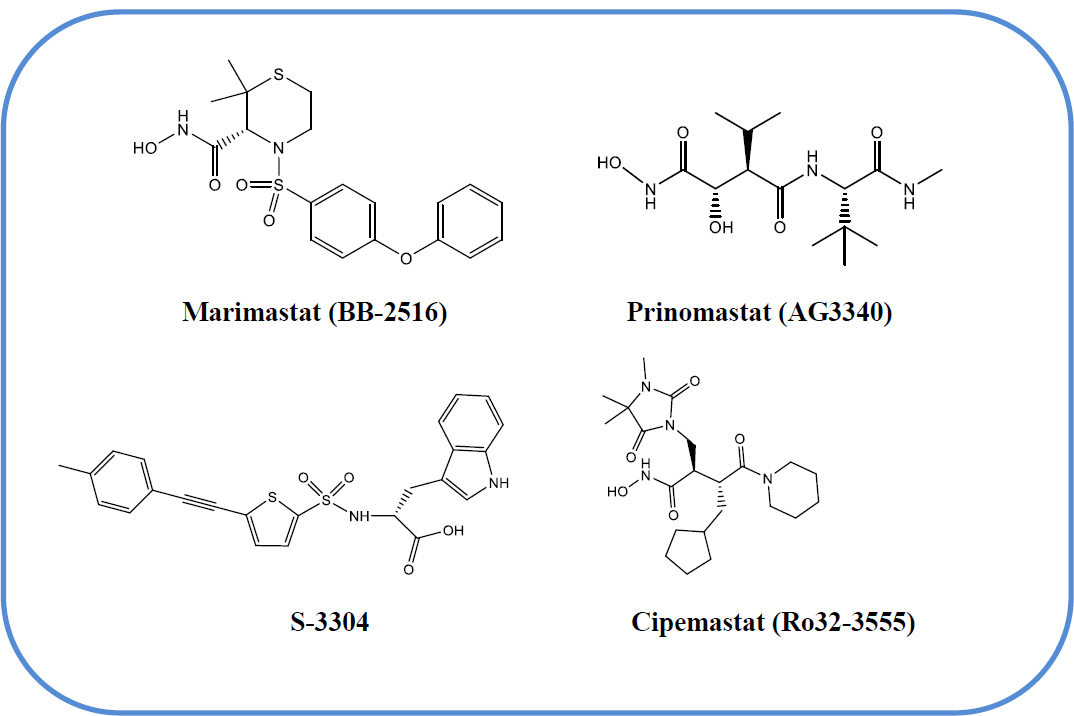
Chemical structures of clinically evaluated MMP inhibitors.
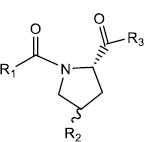 |
||||
| S. No. | R1 | R2 | R3 | IC50 (µM) |
|---|---|---|---|---|
| 1 | CH3 | OH | OCH3 | 2.0 ± 0.2 |
| 2 | CH=CHC6H3(OCH3)2-3',4'(E) | OH | OCH3 | 2.9 ± 0.2 |
| 3 | CH3 | OSO2C6H4CH3-p | OCH3 | 0.3 ± 0.1 |
| 4 | CH=CHC6H3(OCH3)2-3',4'(E) | OSO2CH3 | OCH3 | 1.1 ± 0.04 |
| 5 | CH3 | N3 | OCH3 | 2.6 ± 0.3 |
| 6 | CH=CHC6H3(OCH3)2-3',4'(E) | N3 | OCH3 | 3.4 ± 0.4 |
| 7 | CH3 | NH2 | OCH3 | 1.5 ± 0.1 |
| 8 | CH=CHC6H3(OCH3)2-3',4'(E) | NH2 | OCH3 | ND |
| 9 | CH2CH2C6H3(OCH3)2-3',4' | NH2 | OCH3 | 2.1 ± 0.3 |
| 10 | CH3 | NHCOC6H5 | OCH3 | 2.7 ± 0.3 |
| 11 | CH3 | NHCOC6H4Cl-p | OCH3 | 4.2 ± 0.4 |
| 12 | CH3 | NHCOC6H4Br-p | OCH3 | 6.1 ± 1.0 |
| 13 | CH3 | NHCOC6H4 NO2-p | OCH3 | 5.2 ± 0.6 |
| 14 | CH3 | NHCOC6H2(OCH3)3-3',4',5' | OCH3 | 0.1 ± 0.01 |
| 15 | CH3 | NHCOC5H4 N-3' | OCH3 | 1.0 ± 0.1 |
| 16 | CH3 | NHCOC5H4 N-4' | OCH3 | 2.2 ± 0.2 |
| 17 | CH3 | NHCOC4H3N2-2',5' | OCH3 | 2.1 ± 0.3 |
| 18 | CH3 | NHCOCH=CHC6H5(E) | OCH3 | 0.1 ± 0.01 |
| 19 | CH3 | NHCOCH=CHC6H3(OCH3)2-3',4'(E) | OCH3 | 0.8 ± 0.1 |
| 20 | CH=CHC6H3(OCH3)2-3',4'(E) | NHCOC5H4 N-3' | OCH3 | 3.0 ± 0.04 |
| 21 | CH2CH2C6H3(OCH3)2-3',4' | NHCOC5H4 N-3' | OCH3 | 2.2 ± 0.2 |
| 22 | CH2CH2C6H3(OCH3)2-3',4' | NHCOCH=CHC6H5(E) | OCH3 | 1.6 ± 0.3 |
| 23 | CH3 | NHCOC6H2(OCH3)3-3',4',5' | NHOH | 0.001 ± 0.0001 |
| 24 | CH3 | NHCOC5H4 N-3' | NHOH | 0.005 ± 0.0004 |
| 25 | CH3 | NHCOCH=CHC6H5(E) | NHOH | 0.004 ± 0.0005 |
| 26 | CH2CH2C6H3(OCH3)2-3',4' | NHCOC5H4 N-3' | NHOH | 0.012 ± 0.002 |
| 27 | CH2CH2C6H3(OCH3)2-3',4' | NHCOCH=CHC6H5(E) | NHOH | 0.011 ± 0.001 |
| 28 | CH3 | NHCOC6H2(OCH3)3-3',4',5' | OH | 0.016 ± 0.002 |
| 29 | CH3 | NHCOC6H2(OCH3)3-3',4',5' | NHCH2COOCH3 | 0.109 ± 0.012 |
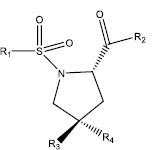 |
|||||
| S. No. | R1 | R2 | R3 | R4 | IC50 (µM) |
|---|---|---|---|---|---|
| 30 | p-CH3C6H4 | OCH3 | OH | H | 0.1 ± 0.02 |
| 31 | C6H5 | OCH3 | OH | H | 0.8 ± 0.1 |
| 32 | CH3 | OCH3 | OH | H | 7.1 ± 0.6 |
| 33 | p-CH3C6H4 | OCH3 | C=O | 0.4 ± 0.04 | |
| 34 | C6H5 | OCH3 | C=O | 2.0 ± 0.1 | |
| 35 | CH3 | OCH3 | C=O | 9.5 ± 1.2 | |
| 36 | p-CH3C6H4 | OCH3 | O–(CH2)2–O | 0.1 ± 0.01 | |
| 37 | p-CH3C6H4 | OCH3 | O–(CH2)3–O | 0.2 ± 0.03 | |
| 38 | p-CH3C6H4 | OCH3 | O–(CH2)4–O | 0.3 ± 0.04 | |
| 39 | C6H5 | OCH3 | O–(CH2)2–O | 0.2 ± 0.04 | |
| 40 | C6H5 | OCH3 | O–(CH2)3–O | 1.1 ± 0.1 | |
| 41 | C6H5 | OCH3 | O–(CH2)4–O | 1.5 ± 0.1 | |
| 42 | p-CH3C6H4 | NHOH | O–(CH2)2–O | 0.003 ± 0.0004 | |
| 43 | p-CH3C6H4 | NHOH | O–(CH2)3–O | 0.006 ± 0.0005 | |
| 44 | p-CH3C6H4 | NHOH | O–(CH2)4–O | 0.008 ± 0.001 | |
| 45 | C6H5 | NHOH | O–(CH2)2–O | 0.008 ± 0.0008 | |
| 46 | C6H5 | NHOH | O–(CH2)3–O | 0.011 ± 0.001 | |
| 47 | C6H5 | NHOH | O–(CH2)4–O | 0.012 ± 0.001 | |
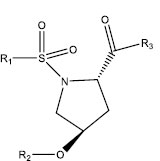 |
||||
| S. No. | R1 | R2 | R3 | IC50 (µM) |
|---|---|---|---|---|
| 48 | p-CH3C6H4 | H | OCH3 | 0.1 ± 0.02 |
| 49 | C6H5 | H | OCH3 | 0.8 ± 0.1 |
| 50 | CH3 | H | OCH3 | 7.1 ± 0.6 |
| 51 | p-CH3C6H4 | C6H5CO | OCH3 | 3.2 ± 0.3 |
| 52 | p-CH3C6H4 | p-ClC6H4CO | OCH3 | 2.2 ± 0.2 |
| 53 | p-CH3C6H4 | p-CH3C6H4SO2 | OCH3 | 0.007 ± 0.001 |
| 54 | p-CH3C6H4 | C6H5SO2 | OCH3 | 0.05 ± 0.006 |
| 55 | p-CH3C6H4 | CH3SO2 | OCH3 | 0.2 ± 0.01 |
| 56 | p-CH3C6H4 | (E)C6H5CH=CHCO | OCH3 | 0.04 ± 0.008 |
| 57 | C6H5 | C6H5CO | OCH3 | 3.5 ± 0.4 |
| 58 | C6H5 | p-ClC6H4CO | OCH3 | 2.7 ± 0.2 |
| 59 | C6H5 | p-CH3C6H4SO2 | OCH3 | 0.04 ± 0.003 |
| 60 | C6H5 | C6H5SO2 | OCH3 | 0.01 ± 0.001 |
| 61 | C6H5 | CH3SO2 | OCH3 | 1.3 ± 0.1 |
| 62 | C6H5 | (E)C6H5CH=CHCO | OCH3 | 0.09 ± 0.01 |
| 63 | CH3 | C6H5CO | OCH3 | 5.2 ± 0.4 |
| 64 | CH3 | p-ClC6H4CO | OCH3 | 1.5 ± 0.2 |
| 65 | CH3 | p-CH3C6H4SO2 | OCH3 | 0.2 ± 0.01 |
| 66 | CH3 | C6H5SO2 | OCH3 | 0.7 ± 0.06 |
| 67 | CH3 | CH3SO2 | OCH3 | 8.9 ± 1.0 |
| 68 | CH3 | (E)C6H5CH=CHCO | OCH3 | 0.5 ± 0.07 |
| 69 | p-CH3C6H4 | H | NHOH | 0.002 ± 0.0002 |
| 70 | C6H5 | H | NHOH | 0.004 ± 0.0003 |
| 71 | CH3 | H | NHOH | 0.02 ± 0.001 |
Pyrrolidine derivatives have garnered significant attention in medicinal chemistry due to their versatile biological activities and favourable pharmacokinetic profiles [21]. In the context of MMP inhibition, the pyrrolidine scaffold is particularly relevant, as it mimics the transition state of peptide substrates and facilitates selective binding to the enzyme's active site [22]. Numerous studies have demonstrated that pyrrolidine-based compounds exhibit potent inhibitory activity against MMP-2, MMP-8, MMP-9, and MMP-14, which are critically involved in tumor invasion, metastasis, and angiogenesis [23-25]. Therefore, the incorporation of pyrrolidine moieties into the design of novel inhibitors represents a rational and effective strategy for anticancer drug development.
The 4-substituted pyrrolidine derivative functionalized with sulfonyl and hydroxamate was chosen for structural and biological reasons. It is widely known that the pyrrolidine core can interact with the catalytic zinc ion in the MMP active site, especially in MMP-2 [26, 27]. Meanwhile, the sulfonyl and hydroxamate groups that are attached improve S1′ pocket binding and zinc chelation [28]. Nitrogen and other substituents were carefully added to the pyrrolidine and to the para-position of the phenyl ring to refine potency, enhance isoform selectivity, and modify steric and electronic characteristics [29]. Prior SAR data and docking investigations further confirmed this design, showing enhanced binding affinity, attractive ADMET profiles, and balanced hydrophobicity and polarity, all of which together make the derivative a promising candidate for selective MMP-targeted anticancer therapy.
This study offers significant value in the early-stage development of anticancer therapeutics by targeting MMP-2, a critical enzyme implicated in tumor invasion and metastasis. The integrated QSAR, molecular docking, and ADMET approach enables the identification and optimization of pyrrolidine-based scaffolds with high selectivity, potency, and favorable pharmacokinetic profiles. Such in silico methods not only reduce the time and cost of drug discovery but also help in prioritizing lead compounds for synthesis and further biological evaluation. The findings contribute to rational drug design strategies and provide a framework for developing next-generation MMP-2 inhibitors as potential anticancer agents. The aim was to obtain a mathematical model that could be used for the prediction of the inhibitory potency of new pyrrolidine derivatives against MMP-2. The data set was subjected to molecular docking along with the evaluation of ADMET properties using in silico tools.
2. MATERIALS AND METHODS
2.1. QSAR Studies
A series of pyrrolidine derivatives acting as MMP-2 inhibitors was reported by Cheng et al. [18-20]. All the compounds of the series, along with their IC50 (µM), are listed in Table 1. The inhibition activity of these compounds and the relevant physicochemical parameters that were found to be correlated with the activity are listed in Table 2 along with their observed and calculated inhibition activity. The activity term IC50 refers to the molar concentration of the compounds leading to 50% inhibition of the enzyme’s activity for MMP-2, which were converted to-logIC50 (pIC50) values. The relevant physicochemical parameters that were found to be useful in the correlation were the molar refractivity (CMR) of the compounds and the calculated hydrophobicity (ClogP) of the compounds. Both the parameters have been calculated using ChemDraw (15.0 version) and Chemsketch (23.0 version). In addition, some indicator variables were also used, and these are indicated by I1 and I2. These indicator variables described the specific roles of certain segments of the molecules.
2.2. Molecular Docking Studies
We selected the catalytic domain of human MMP-2 (PDB ID: 1HOV) as our target protein. This high-resolution crystal structure was chosen because it contains the functionally active site with the essential catalytic zinc ion, which is the precise location where inhibitory compounds exert their effect [30]. The presence of a co-crystallized hydroxamate inhibitor [31] provides a crucial reference point for validating our docking protocol and serves as a benchmark for evaluating the binding modes and affinities of our designed pyrrolidine derivatives [32, 33].
2.2.1. Ligand Preparation
The 2D structures of the designed ligands were sketched using ChemDraw Professional (v16.0.0.82) [34]. These structures were saved in .sdf format and subsequently subjected to energy minimization using Chem3D 16.0 software. The MM2 force field was applied with a minimum RMS gradient of 0.010 to relieve any steric clashes and to optimize the molecular geometry [35, 36]. The energy-minimized structures were then converted into pdbqt format using Open Babel GUI software [37]. Ligand files were prepared for docking using AutoDock Tools 1.5.7 [38].
2.2.2. Discovery Studio
Discovery Studio (BIOVIA) was utilized for receptor and ligand preparation by removing water molecules and non-target residues. It was also employed to visualize the 2D and 3D interactions between the ligands and the target proteins, thereby facilitating evaluation of ligand–protein binding strength [39].
2.2.3. Preparation of Target Protein
The 3D crystal structure of MMP-2 was retrieved from the Protein Data Bank (PDB ID: 1HOV), which corresponds to the solution structure of the catalytic domain of MMP-2 complexed with SC-74020 [40]. Among the 11 macromolecular models present, Model 1, comprising the catalytic domains S1', S1, S2', and S3', was selected for docking, based on literature evidence of minimal structural variation among models [41]. Receptor preparation included removal of water molecules and bound ligands, addition of polar hydrogens, and assignment of Kollman charges using BIOVIA Discovery Studio Visualizer 2021 [39, 42, 43].
| Compound | CMR | CLogP | I1 | I2 | log (1/IC50) | ||
|---|---|---|---|---|---|---|---|
| Obsda | Calcd | LOOd | |||||
| 1 | 4.460 | -0.980 | 0 | 0 | 5.700 | 5.685 | 5.682 |
| 2 | 8.940 | 0.970 | 0 | 0 | 5.540 | 5.920 | 5.937 |
| 3b | 8.300 | 1.150 | 0 | 0 | 6.520 | 5.794 | - |
| 4b | 10.280 | 1.360 | 0 | 0 | 5.960 | 6.027 | - |
| 5 | 5.050 | 0.760 | 0 | 0 | 5.590 | 5.421 | 5.402 |
| 6 | 9.540 | 2.710 | 0 | 0 | 5.470 | 5.657 | 5.67 |
| 7 | 4.670 | -0.910 | 0 | 0 | 5.820 | 5.700 | 5.686 |
| 9 | 8.880 | 0.850 | 0 | 0 | 5.680 | 5.936 | 5.947 |
| 10 | 7.680 | 0.680 | 0 | 0 | 5.570 | 5.803 | 5.808 |
| 11 | 8.170 | 1.590 | 0 | 0 | 5.380 | 5.690 | 5.697 |
| 12 | 8.460 | 1.740 | 0 | 0 | 5.210 | 5.700 | 5.719 |
| 13 | 8.290 | 0.850 | 0 | 0 | 5.280 | 5.854 | 5.874 |
| 14 | 9.530 | 0.160 | 0 | 0 | 7.000 | 6.164 | 6.064 |
| 15b | 7.470 | -0.070 | 0 | 0 | 6.000 | 5.921 | - |
| 16 | 7.470 | -0.070 | 0 | 0 | 5.660 | 5.923 | 5.936 |
| 17 | 7.260 | -0.510 | 0 | 0 | 5.680 | 5.981 | 6.008 |
| 18c | 8.890 | 1.580 | 0 | 0 | 7.000 | 5.790 | - |
| 19b | 10.120 | 1.240 | 0 | 0 | 6.100 | 6.029 | - |
| 20 | 11.960 | 1.880 | 0 | 0 | 5.520 | 6.159 | 6.241 |
| 21 | 11.680 | 1.680 | 0 | 0 | 5.660 | 6.160 | 6.228 |
| 22 | 13.100 | 3.330 | 0 | 0 | 5.800 | 6.029 | 6.068 |
| 23 | 9.440 | -1.290 | 1 | 0 | 9.000 | 8.342 | 8.237 |
| 24 | 7.380 | -1.520 | 1 | 0 | 8.300 | 8.101 | 8.079 |
| 25 | 8.790 | 0.130 | 1 | 0 | 8.400 | 7.969 | 7.915 |
| 26 | 11.590 | 0.230 | 1 | 0 | 7.920 | 8.339 | 8.418 |
| 27 | 13.000 | 1.890 | 1 | 0 | 7.960 | 8.205 | 8.269 |
| 28c | 9.070 | -0.100 | 0 | 0 | 7.800 | 6.150 | - |
| 29 | 10.870 | -0.400 | 0 | 0 | 6.960 | 6.461 | 6.280 |
| 30b | 7.340 | 1.230 | 0 | 1 | 7.000 | 6.360 | - |
| 31 | 6.880 | 0.730 | 0 | 0 | 6.100 | 5.681 | 5.661 |
| 32 | 4.830 | -0.710 | 0 | 0 | 5.150 | 5.683 | 5.732 |
| 33b | 7.220 | 1.900 | 0 | 1 | 6.400 | 6.210 | - |
| 34 | 6.760 | 1.400 | 0 | 0 | 5.700 | 5.531 | 5.520 |
| 35 | 4.710 | 0.110 | 0 | 0 | 5.020 | 5.503 | 5.551 |
| 36 | 8.240 | 1.580 | 0 | 1 | 7.000 | 6.417 | 6.314 |
| 37b | 8.710 | 1.350 | 0 | 1 | 6.700 | 6.527 | - |
| 38 | 9.170 | 1.910 | 0 | 1 | 6.520 | 6.481 | 6.475 |
| 39 | 7.780 | 1.080 | 0 | 0 | 6.700 | 5.737 | 5.705 |
| 40b | 8.240 | 0.850 | 0 | 0 | 5.960 | 5.845 | - |
| 41b | 8.710 | 1.410 | 0 | 0 | 5.820 | 5.799 | - |
| 42 | 8.150 | 0.130 | 1 | 1 | 8.520 | 8.596 | 8.616 |
| 43 | 8.610 | -0.100 | 1 | 1 | 8.220 | 8.706 | 8.813 |
| 44 | 9.080 | 0.460 | 1 | 1 | 8.100 | 8.660 | 8.773 |
| 45 | 7.690 | -0.370 | 1 | 0 | 8.100 | 7.916 | 7.894 |
| 46 | 8.150 | -0.600 | 1 | 0 | 7.960 | 8.026 | 8.033 |
| 47b | 8.610 | -0.040 | 1 | 0 | 7.920 | 7.977 | - |
| 48b | 7.340 | 1.230 | 0 | 1 | 7.000 | 6.360 | - |
| 49 | 6.880 | 0.730 | 0 | 0 | 6.100 | 5.681 | 5.661 |
| 50 | 4.830 | -0.710 | 0 | 0 | 5.150 | 5.683 | 5.732 |
| 51b | 10.350 | 3.860 | 0 | 1 | 5.490 | 6.256 | - |
| 52 | 10.840 | 4.570 | 0 | 1 | 5.660 | 6.184 | 6.329 |
| 53c | 11.190 | 3.370 | 0 | 1 | 8.150 | 6.470 | - |
| 54 | 10.720 | 2.870 | 0 | 1 | 7.300 | 6.506 | 6.361 |
| 55 | 8.680 | 1.630 | 0 | 1 | 6.700 | 6.469 | 6.428 |
| 56c | 11.560 | 4.310 | 0 | 1 | 7.400 | 6.334 | - |
| 57 | 9.890 | 3.360 | 0 | 0 | 5.460 | 5.577 | 5.593 |
| 58 | 10.380 | 4.070 | 0 | 0 | 5.570 | 5.504 | 5.486 |
| 59c | 10.720 | 2.870 | 0 | 0 | 7.400 | 5.788 | - |
| 60c | 10.260 | 2.370 | 0 | 0 | 8.000 | 5.824 | - |
| 61b | 8.210 | 1.130 | 0 | 0 | 5.890 | 5.785 | - |
| 62c | 11.090 | 3.810 | 0 | 0 | 7.050 | 5.652 | - |
| 63 | 7.840 | 1.910 | 0 | 0 | 5.280 | 5.580 | 5.595 |
| 64 | 8.330 | 2.620 | 0 | 0 | 5.820 | 5.507 | 5.480 |
| 65 | 8.680 | 1.420 | 0 | 0 | 6.700 | 5.794 | 5.768 |
| 66 | 8.210 | 0.920 | 0 | 0 | 6.150 | 5.829 | 5.817 |
| 67 | 6.170 | -0.320 | 0 | 0 | 5.050 | 5.791 | 5.839 |
| 68 | 8.820 | 2.650 | 0 | 0 | 6.300 | 5.569 | 5.507 |
| 69b | 7.250 | -0.220 | 1 | 1 | 8.700 | 8.540 | - |
| 70 | 6.780 | -0.720 | 1 | 0 | 8.400 | 7.859 | 7.790 |
| 71 | 4.730 | -2.160 | 1 | 0 | 7.700 | 7.860 | 7.889 |
2.2.4. Docking Studies
Molecular docking studies were conducted using AutoDock Vina 1.1.2 [44], aimed at identifying selective ligands for MMP-2. Docking simulations were carried out on a Windows-based PC equipped with an Intel Core i3 processor, 8 GB RAM, and a 64-bit operating system [45]. The protein file was converted to pdbqt format using AutoDock Tools 1.5.7 [46]. A grid box was generated with dimensions of 100 Å × 100 Å × 100 Å, centered at coordinates X = 8.572, Y = 20.228, Z = 12.664 Å, using a default grid spacing of 0.375 Å. Docking parameters included a maximum of 2,500,000 energy evaluations using the Lamarckian Genetic Algorithm (LGA) as the search engine, with all other settings kept at default values [47]. 2D and 3D visualizations of ligand-protein interactions were performed using Discovery Studio to analyze binding poses and interaction profiles. The docking results were evaluated based on the lowest binding energy (ΔG) scores within the most populated conformational cluster [48].
2.2.5. ADMET Studies
In silico ADMET prediction studies to check the lipophilicity, solubility, pharmacokinetics, and drug likeness, etc. properties were carried out using SwissADME from the Swiss Institute of Bioinformatics [35].
3. RESULTS AND DISCUSSION
The data set for MMP-2 inhibition activity was divided into a training set as well as test set. Compound 8 is not included in any data set due to the unavailability of its IC50. The data of the test set for the activity are given in bold. The compounds for the test set were selected arbitrarily, taking into consideration the wide structural diversity and span in their activity data. When a multiple regression analysis was performed on the training set, which revealed highly significant correlations as shown by Eqs. (1-4) were obtained.
In Eqs. (1-4), n is the number of data points, r is the correlation coefficient of the regression, r2cv is the square of cross-validated correlation coefficient obtained by leave-one-out (LOO) jackknife procedure, s is the standard deviation, and F is the ratio between the variances of calculated and observed activities (within parenthesis the figure refers to the F value at 99% level). The data with ± sign within the parentheses refer to 95% confidence intervals for the coefficients of the variables as well as for the intercept. The value of r2cv is calculated to find the validity of the correlation. The r2cv is defined as Eq. (5):
where yi,obsd, and yi,cald are the observed and calculated (from LOO) activity values of compound i, respectively, and ȳobsd is the average of the observed activities of all compounds used in the correlation. The correlation is supposed to be valid if r2cv> 0.60. From this point of view, our correlation is quite valid. However, the predictive ability of any correlation equation is measured by using it to predict the activity of the compounds in the test set and calculating the value of r2pred, which is defined as Eq (6):
where yi, pred is the predicted activity of compound i. A value of r2pred> 0.5 signifies a good predictive ability of the correlation equation. For equation (1), r2pred values are as high as 0.798. The activity values predicted from these equations for MMP-2 inhibition activity, for the compounds in the respective test sets, are given (in bold) in Table 2. A comparison shows these predicted values are in very good agreement with the corresponding observed ones. In the training set, the calculated values are also found to be in excellent agreement with the observed ones. All these observations can be better visualized in the graphs drawn between the predicted and observed activities (Fig. 2).
Eq. (1) suggests that for the inhibition of MMP-2, CMR plays a pivotal role in influencing the biological activity of compounds, including their potential. This descriptor reflects molecular size, polarizability, and electronic properties, which are critical for effective target interactions. A positive coefficient of the CMR parameter in QSAR models suggests that molecular size and polarizability contribute positively towards inhibitory properties. Interestingly, while increased molar refractivity often correlates with larger molecular size and enhanced van der Waals interactions or steric complementarity, certain contexts highlight steric effect where smaller molecules exhibit improved target binding. Additionally, the degree of unsaturation in compounds significantly influences molar refractivity, with unsaturated molecules demonstrating superior activity compared to saturated ones, likely due to enhanced electronic and structural compatibility with the target. Overall, the CMR descriptor's positive contribution to pIC50 values underscores its importance in guiding the rational design of effective and potent compounds by balancing size, polarizability, and structural features. This makes it a crucial parameter in developing molecules with optimized biological efficacy.
The second parameter is the ClogP descriptor, which represents the logarithm of the partition coefficient (logP) between octanol and water, i.e., hydrophobicity. A negative coefficient of ClogP in a QSAR model indicates that increased hydrophobicity negatively impacts the biological activity of the compound. This suggests that excessively hydrophobic molecules may have reduced solubility in aqueous environments, limiting their bioavailability or ability to interact effectively with the target receptor. To solve this problem, optimizing the hydrophilic-lipophilic balance by reducing hydrophobicity (lowering ClogP values) may enhance the compound's biological activity. Thus, compounds with moderate or lower hydrophobicity are likely more effective in achieving the desired activity in this context.
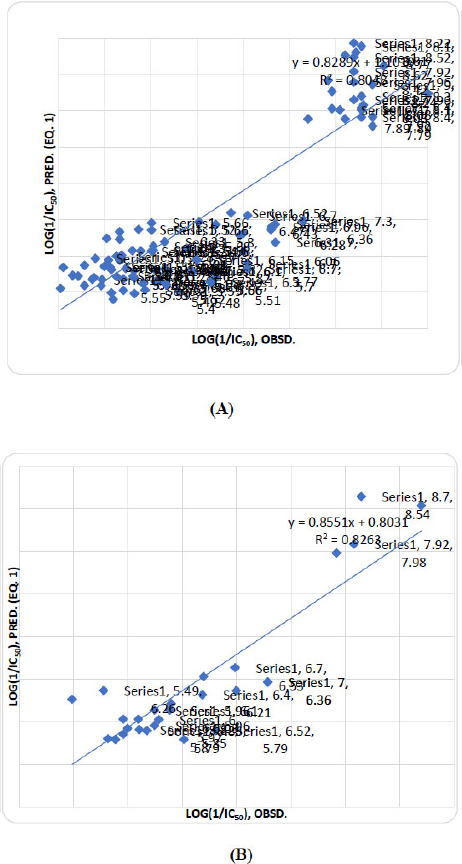
Graph of observed activity vs predicted activity (Eq. 1) of training (A) and test (B) set compounds.
All the QSAR models developed in this study incorporate indicator parameters, denoted as I1 and I2, to represent specific structural features of molecules. These parameters are binary variables, with a value of 1 indicating the presence of the corresponding structural feature and 0 indicating its absence. This approach allows the models to effectively encode and evaluate the contribution of these features to the biological activity of the compounds. In this case:
I1 corresponds to the presence of an (S)-N-hydroxy-2-carboxamide group, with a value of 1 indicating its presence. I2 corresponds to the presence of a 1-tosyl aromatic ring group, with a value of 1 indicating its presence. The positive coefficients for both I1 and I2 indicate that these functional groups significantly enhance the biological activity of the compounds. Specifically, (S)-N-hydroxy-2-carboxamide group (I1) likely contributes to activity through critical interactions such as hydrogen bonding with the target MMP-2, enhancing the compound's affinity and potency. The 1-tosyl aromatic ring group (I2) may play a role in stabilizing interactions, such as π-π stacking or hydrophobic contacts, further improving target binding and overall efficacy. Together, these features contribute synergistically to the biological activity, suggesting that compounds designed with both the (S)-N-hydroxy-2-carboxamide group and the 1-tosyl aromatic ring group are more likely to exhibit enhanced performance. These structural insights can be helpful to guide the rational design of new compounds for improved activity.
Eq. ( 2 ) demonstrates that this QSAR model is an excellent and highly predictive tool for understanding the structural requirements for potent inhibitors. The model demonstrates a very strong correlation ( r = 0.893) and excellent predictive power, as indicated by the cross-validated value ( r 2 cv = 0.796). High F -statistic (58.733) confirms the model is statistically significant overall. In this model, the CMR descriptor, which reflects molecular volume and polarizability, contributes positively (+0.121), indicating that moderately bulky and polarizable molecules enhance binding affinity, likely due to better interactions within the active site. The ClogP descriptor has a small negative coefficient (-0.110), indicating that greater lipophilicity may marginally reduce MMP-2 inhibitory activity, possibly due to impaired solubility or decreased target engagement. This effect is also statistically insignificant, as reflected by its relatively large standard error (±0.173). In contrast, the indicator variable I 1 shows a large, statistically significant positive coefficient (+2.132 ± 0.473), emphasizing that the presence of a specific structural motif plays a critical role in enhancing MMP-2 inhibition. Among all descriptors, I 1 is the most influential, providing a clear direction for structural optimization.
In conclusion, while CMR and ClogP contribute marginally, the structural feature denoted by I 1 is the principal determinant of MMP-2 inhibitory potency. This insight offers a strategic direction for rational drug design focusing on incorporating structural elements linked to I 1 to enhance biological efficacy. Overall, this model is highly valuable for rational design and screening of MMP-2 inhibitors, especially those containing the feature represented by I 1 , such as specific substitutions on a pyrrolidine scaffold.
Eq. ( 3 ) is obtained by rearranging the parameters in a combination of CMR, ClogP, and I 2 , and then correlating this combination with the observed inhibition activity for MMP-2. In this equation, statistical parameters show a good model fit ( r = 0.756) and acceptable predictive power (cross-validated r 2 cv = 0.571), with a standard error of ( s = 0.787) and an F-value of 19.958, indicating statistical significance. Each descriptor contributes meaningfully to the model. A positive coefficient for CMR (0.349) indicates that increasing a compound's bulk and polarizability enhances potency, suggesting a role for favorable van der Waals interactions. Conversely, a negative coefficient for ClogP (-0.671) shows that increasing lipophilicity is detrimental to activity, implying an optimal range is required to avoid poor solubility or non-specific binding. Most significantly, the large positive coefficient for the indicator variable I 2 (1.231) highlights a specific structural feature as a major potency-enhancing group. Overall, the model suggests that for improved MMP-2 inhibitory activity, compounds should have moderate size, good polarizability, optimized topological features, and balanced hydrophilicity. The inclusion of the -logIC 50 transformation aligns higher values with higher potency, facilitating clear interpretation. This model provides a reliable foundation for guiding the design of novel pyrrolidine-based MMP-2 inhibitors with potential anticancer applications and supports rational lead optimization through molecular descriptor tuning.
However, Eq. ( 4 ) statistically, the model shows a moderate correlation ( r = 0.651) and acceptable internal predictivity ( r 2 cv = 0.424), with a standard error ( s ) of 0.902 and a significant F -value of 16.906, indicating the model is not due to chance. Descriptor analysis reveals that a positive coefficient for CMR (0.362), representing molecular size and polarizability, contributes positively, suggesting that compounds with greater volume or polarizability tend to be more potent inhibitors. In contrast, ClogP (-0.610) shows a negative contribution, indicating that highly lipophilic compounds may have reduced activity, likely due to solubility issues or nonspecific binding.
Overall, the model provides insights into the structural features favoring activity and can guide the design of more effective MMP-2 inhibitors. Specifically, optimizing molecular size while maintaining moderate lipophilicity may enhance the potency. While the model is useful for early-stage screening, further refinement with additional descriptors or compounds could improve predictive power for lead optimization ( Table 3 ).
3.1. Design of New Inhibitors and Interpretation of Model Descriptors
The four molecular descriptors presented in the QSAR model provide a guide for the design of new derivatives with increased MMP-2 inhibition activity, which are of great importance for improving activity. The relative significance of the descriptors presented in the model was determined on the basis of their standardized regression coefficients, as shown in Eq. (1). The standardized regression coefficients for each descriptor provide important information about the effect of the molecular descriptors and their degree of contribution to the model developed. The contributing parameters are as follows: CMR, ClogP, I1 ((S)-N-hydroxy-2-carboxamide group) and I2 (1-tosyl group).
To explore molecular variations, we selected the molecule with the highest level of MMP-2 inhibition activity as the reference. Structural modifications were then performed by substituting atoms and atomic groups at various sites in the molecule. Using the optimal QSAR model, pIC50 values of the modified compounds were estimated based on Eq. (1), and their leverage values were calculated to determine whether the compounds fell within the domain of applicability. This process resulted in the design of 20 new molecules.
Molecular docking and ADMET prediction studies were conducted on the 20 designed molecules to evaluate their potential as biologically active, potent, and safe candidates. The combined outcomes of QSAR modeling, molecular docking, and ADMET analyses indicated that all selected compounds exhibited favourable biological profiles and promising pharmacological activity, as summarized in Table 4. The predicted pIC50 values were calculated using Eq. (1), and leverage values were determined to confirm that the compounds fell within the defined applicability domain of the QSAR model.
MMP-2 inhibitory activity of all 20 molecules screened out is higher than the reported reference compounds 23, 42, 69 at the reference value that corresponds to (pIC50 = 9.0, 8.52 and 8.70). The in silico MMP-2 inhibitory activity and mechanism of action of the designed data set were predicted through docking studies.
| - | CMR | CLogP | I1 | I2 |
|---|---|---|---|---|
| CMR | 1.000 | -0.714 | -0.508 | 0.108 |
| CLogP | - | 1.000 | 0.672 | -0.310 |
| I1 | - | - | 1.000 | -0.294 |
| I2 | - | - | - | 1.000 |
| S. No. | Compound Structure (SMILES Notation) | CMR | CLogP | I1 | I2 |
Log (1/IC50) |
Docking Score (kcal/mol) |
|---|---|---|---|---|---|---|---|
| M1 | O=S(=O)(N1CC(CC1C(=O)NO)C(=O)NC1CCN(CC1)OC)c1ccc(C)cc1 | 11.088 | -0.236 | 1 | 1 | 9.076 | -8.1 |
| M2 | O=S(=O)(N1CC(CC1C(=O)NO)C(=O)NC1CCN(C)CC1)c1ccc(C)cc1 | 10.935 | -0.227 | 1 | 1 | 9.053 | -8.2 |
| M3 | O=S(=O)(N1CC(CC1C(=O)NO)C(=O)NC1CCN(CC)CC1)c1ccc(C)cc1 | 11.399 | 0.302 | 1 | 1 | 9.012 | -8.5 |
| M4 | O=S(=O)(N1CC(CC1C(=O)NO)C(=O)NC1CCN(CC1)C(C)C)c1ccc(C)cc1 | 11.863 | 0.611 | 1 | 1 | 9.015 | -7.6 |
| M5 | CC(C)(C)N1CCC(CC1)NC(=O)C1CN(C(C1)C(=O)NO)S(=O)(=O)c1ccc(C)cc1 | 12.326 | 0.84 | 1 | 1 | 9.034 | -7.7 |
| M6 | O=S(=O)(N1CC(CC1C(=O)NO)C(=O)NN1CCN(C)CC1)c1ccc(C)cc1 | 10.84 | 0.652 | 1 | 1 | 8.865 | -8.3 |
| M7 | O=S(=O)(N1CC(CC1C(=O)NO)C(=O)NN1CCN(CC1)CC)c1ccc(C)cc1 | 11.304 | 1.181 | 1 | 1 | 8.824 | -8.1 |
| M8 | O=S(=O)(N1CC(CC1C(=O)NO)C(=O)NN1CCN(CC1)C(C)C)c1ccc(C)cc1 | 11.768 | 1.49 | 1 | 1 | 8.827 | -8.2 |
| M9 | CC(C)(C)N1CCN(NC(=O)C2CN(C(C2)C(=O)NO)S(=O)(=O)c2ccc(C)cc2)CC1 | 12.231 | 1.77 | 1 | 1 | 8.836 | -8.4 |
| M10 | O=S(=O)(N1CC(CC1C(=O)NO)C(=O)N1CCC(CC1)C(F)(F)F)c1ccc(C)cc1 | 10.613 | 1.072 | 1 | 1 | 8.75 | -8.9 |
| M11 | COc1c(OC)cc(NC2CC(N(C2)S(=O)(=O)c2ccc(C)cc2)C(=O)NO)cc1OC | 11.823 | 1.448 | 1 | 1 | 8.843 | -8.1 |
| M12 | COc1cc(ccc1OC)NC1CN(C(C1)C(=O)NO)S(=O)(=O)c1ccc(C)cc1 | 11.206 | 1.835 | 1 | 1 | 8.68 | -8.3 |
| M13 | O=S(=O)(N1CC(OC2CCN(CC2)OC)CC1C(=O)NO)c1ccc(C)cc1 | 10.373 | 0.45 | 1 | 1 | 8.84 | -7.1 |
| M14 | O=S(=O)(N1CC(NC2CCN(CC2)OC)CC1C(=O)NO)c1ccc(C)cc1 | 10.589 | 0.195 | 1 | 1 | 8.921 | -8.2 |
| M15 | O=S(=O)(N1CC(NC2CCN(C)CC2)CC1C(=O)NO)c1ccc(C)cc1 | 10.436 | 0.204 | 1 | 1 | 8.898 | -8 |
| M16 | O=S(=O)(N1CC(Nc2ccc(N)cc2)CC1C(=O)NO)c1ccc(C)cc1 | 10.341 | 0.821 | 1 | 1 | 8.762 | -7.8 |
| M17 | O=S(=O)(N1CC(CC1C(=O)NO)C(=O)Nc1ccc(cc1)OC)c1ccc(C)cc1 | 11.089 | 1.754 | 1 | 1 | 8.68 | -8.9 |
| M18 | O=S(=O)(N1CC(CC1C(=O)NO)NC(=O)N1CCC(C)=N1)c1ccc(C)cc1 | 10.351 | -0.075 | 1 | 1 | 8.942 | -8.2 |
| M19 | O=S(=O)(N1CC(CC1C(=O)NO)NC(=O)N1CCC(Cl)=N1)c1ccc(C)cc1 | 10.378 | 1.231 | 1 | 1 | 8.686 | -8.3 |
| M20 | O=S(=O)(N1CC(CC1C(=O)NO)NC(=O)N1CCC(Br)=N1)c1ccc(C)cc1 | 10.664 | 1.431 | 1 | 1 | 8.686 | -8.3 |
| Compound | Binding Affinity Score |
|---|---|
| 23 | -6.6 |
| 42 | -8.2 |
| 69 | -6.6 |
3.2. Docking Results and Analysis
The designed pyrrolidine derivatives were docked against the MMP-2 target, and their binding affinities were compared to known inhibitors (e.g., compounds 23, 42, and 69). Compound 42 was redocked to validate the docking protocol (Fig. 3). Among the 20 designed compounds, several exhibited superior docking scores compared to reference inhibitors. Notably, compounds M3, M6, M9, M10, M12, M17, M19, and M20 displayed significantly enhanced binding affinities, with ΔG values of -8.5, -8.3, -8.4, -8.9, -8.3, -8.9, -8.3, and -8.3 kcal/mol, respectively. These compounds interacted with key amino acid residues within the active site, including Leu A:83, His A:120, His A:130, Ala A:84, Glu A:121, Tyr A:142, Val A:117, Pro A:140, Leu A:82, Tyr A:74, and His A:85.
Figure 3 illustrates the docked poses of the reference compound (42), highlighting its strong binding affinity toward MMP-2. Furthermore, compounds M28 and M46 were identified as top binders, both demonstrating a minimum ΔG binding score of -8.9 kcal/mol. These results suggest that both of them form the most stable complexes with MMP-2 and represent potential lead compounds (Fig. 4a–b) (Tables 5 and 6).
| Compound | Binding Affinity Score (kcal/mol) | Compound | Binding Affinity Score (kcal/mol) |
|---|---|---|---|
| M1 | -8.1 | M11 | -8.1 |
| M2 | -8.2 | M12 | -8.3 |
| M3 | -8.5 | M13 | -7.1 |
| M4 | -7.6 | M14 | -8.2 |
| M5 | -7.7 | M15 | -8 |
| M6 | -8.3 | M16 | -7.8 |
| M7 | -8.1 | M17 | -8.9 |
| M8 | -8.2 | M18 | -8.2 |
| M9 | -8.4 | M19 | -8.3 |
| M10 | -8.9 | M20 | -8.3 |
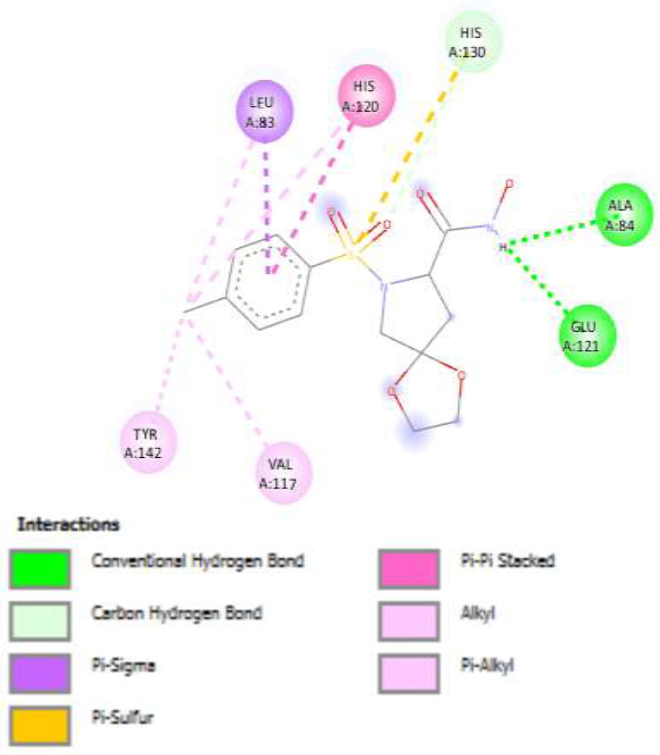
Binding interactions for reference compound (42).
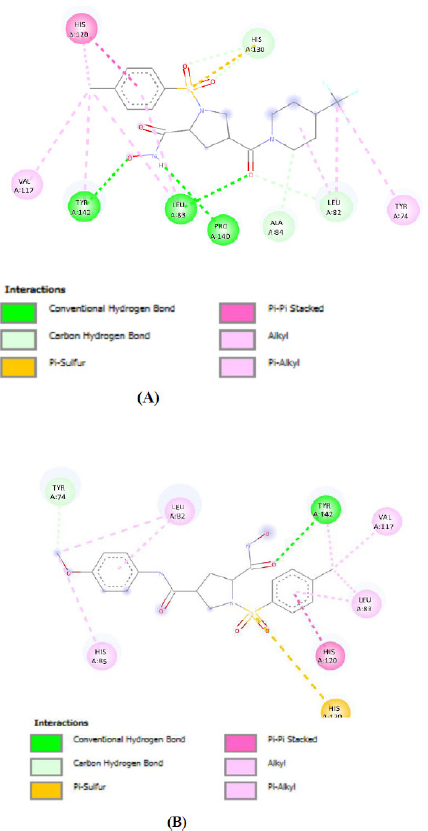
Binding interaction of the most active designed molecules (A) M28 and (B) M46.
3.3. Drug Likeness and in silico ADMET Prediction
The SwissADME web tool offers free access to a comprehensive suite of efficient and reliable predictive models for evaluating the physicochemical properties, pharmacokinetics, drug-likeness, and medicinal chemistry friendliness of compounds [49-51]. It includes advanced proprietary methods such as BOILED-Egg, iLOGP, and bioavailability radar [52]. These tools are particularly valuable in early drug discovery, where in silico ADME prediction helps to mitigate the time and expense of laboratory trials, reducing the likelihood of potential drug candidates failing during clinical trials [53]. An ideal drug candidate is expected to exhibit rapid absorption, effective distribution, efficient metabolism, and safe elimination without adverse effects. The Lipinski rule of five, the most widely recognized guideline for drug-likeness, defines acceptable limits for four key molecular properties of orally active compounds: molecular weight ≤500, logP ≤5, hydrogen bond acceptors (HBA) ≤10, and hydrogen bond donors (HBD) ≤5 [54, 55]. Compounds that violate no more than two of these criteria are considered suitable for oral administration [56].
In this study, the drug-likeness and ADMET properties of the proposed compounds were evaluated using the SwissADME tool, as shown in Table 7. All 20 proposedcompounds adhered to Lipinski's rule of five, indicating their potential as orally active candidates. The pharmacokinetic parameters, including absorption, distribution, metabolism, and excretion, were analyzed alongside toxicity predictions to ensure the safety of the candidates [57].
The BOILED-Egg model (Brain or Intestinal Estimated permeation method) is a graphical predictive tool used to evaluate the drug-likeness and oral bioavailability of small molecules [58] based on key physicochemical descriptors, lipophilicity (WLOGP), and topological polar surface area (TPSA) [59]. In this study, the BOILED-Egg model was employed to assess the likelihood of the designed pyrrolidine-based MMP-2 inhibitors to cross the blood-brain barrier (BBB) and/or be absorbed through the human gastrointestinal tract (HGI) [60]. This analysis is crucial because, for anticancer agents targeting MMP-2, especially those intended for systemic or oral administration, adequate intestinal absorption without undesirable CNS penetration is generally preferred [61, 62]. The model helped us identify compounds with favorable permeability and bioavailability profiles, thereby supporting their drug-likeness and therapeutic potential in anticancer applications [63, 64].
The BOILED-Egg model, a unique feature of SwissADME, provides visual predictions of a compound’s gastrointestinal absorption (HIA) and blood–brain barrier (BBB) permeability [65]. Molecules appearing in the “yolk” of the egg represent those with high BBB penetration, while those in the “white” indicate high intestinal absorption [66]. In this study, none of the 20 compounds appeared in the yolk, suggesting no BBB penetration, but all exhibited high gastrointestinal absorption, as shown in Fig. (5). Moreover, the role of P-glycoprotein (PGP) in drug bioavailability was assessed. PGP acts as a drug efflux pump, limiting drug absorption in the gastrointestinal tract and brain while facilitating drug elimination [67]. Molecules identified as PGP substrates (PGP+) are actively pumped out, reducing their bioavailability. As depicted in Fig. (5), blue molecules were predicted as PGP substrates, while red molecules were non-substrates.
| Compound | Molecular Weight | Rotatable Bonds | H-bond Acceptors | H-bond Donors | TPSA (Å2) | LogP | Lipinski’s Violations | Veber Violations |
|---|---|---|---|---|---|---|---|---|
| M1 | 440.51 | 8 | 8 | 3 | 136.66 | 2.11 | 0 | 0 |
| M2 | 424.51 | 7 | 7 | 3 | 127.43 | 2.08 | 0 | 0 |
| M3 | 438.54 | 8 | 7 | 3 | 127.43 | 1.41 | 0 | 0 |
| M4 | 452.57 | 8 | 7 | 3 | 127.43 | 2.78 | 0 | 0 |
| M5 | 466.59 | 8 | 7 | 3 | 127.43 | 2.8 | 0 | 0 |
| M6 | 425.5 | 7 | 8 | 3 | 130.67 | 1.63 | 0 | 0 |
| M7 | 439.53 | 8 | 8 | 3 | 130.67 | 2.49 | 0 | 0 |
| M8 | 453.56 | 8 | 8 | 3 | 130.67 | 2.28 | 0 | 0 |
| M9 | 467.58 | 8 | 8 | 3 | 130.67 | 1.89 | 0 | 0 |
| M10 | 463.47 | 7 | 9 | 2 | 115.4 | 2.06 | 0 | 0 |
| M11 | 465.52 | 9 | 8 | 3 | 134.81 | 2.89 | 0 | 0 |
| M12 | 435.49 | 8 | 7 | 3 | 125.58 | 2.4 | 0 | 0 |
| M13 | 413.49 | 7 | 8 | 2 | 116.79 | 2.24 | 0 | 0 |
| M14 | 412.5 | 7 | 8 | 3 | 119.59 | 1.87 | 0 | 0 |
| M15 | 396.5 | 6 | 7 | 3 | 110.36 | 2.41 | 0 | 0 |
| M16 | 390.46 | 6 | 5 | 4 | 133.14 | 1.82 | 0 | 0 |
| M17 | 433.48 | 8 | 7 | 3 | 133.42 | 2.31 | 0 | 0 |
| M18 | 409.46 | 7 | 7 | 3 | 139.79 | 2.54 | 0 | 0 |
| M19 | 429.88 | 7 | 7 | 3 | 139.79 | 2.08 | 0 | 0 |
| M20 | 474.33 | 7 | 7 | 3 | 139.79 | 0.37 | 0 | 0 |
| Ref. (23) | 381.38 | 9 | 7 | 3 | 126.43 | 1.76 | 0 | 0 |
| Ref. (42) | 342.37 | 4 | 7 | 2 | 113.55 | 1.54 | 0 | 0 |
| Ref. (69) | 300.33 | 4 | 6 | 3 | 115.32 | 0.94 | 0 | 0 |
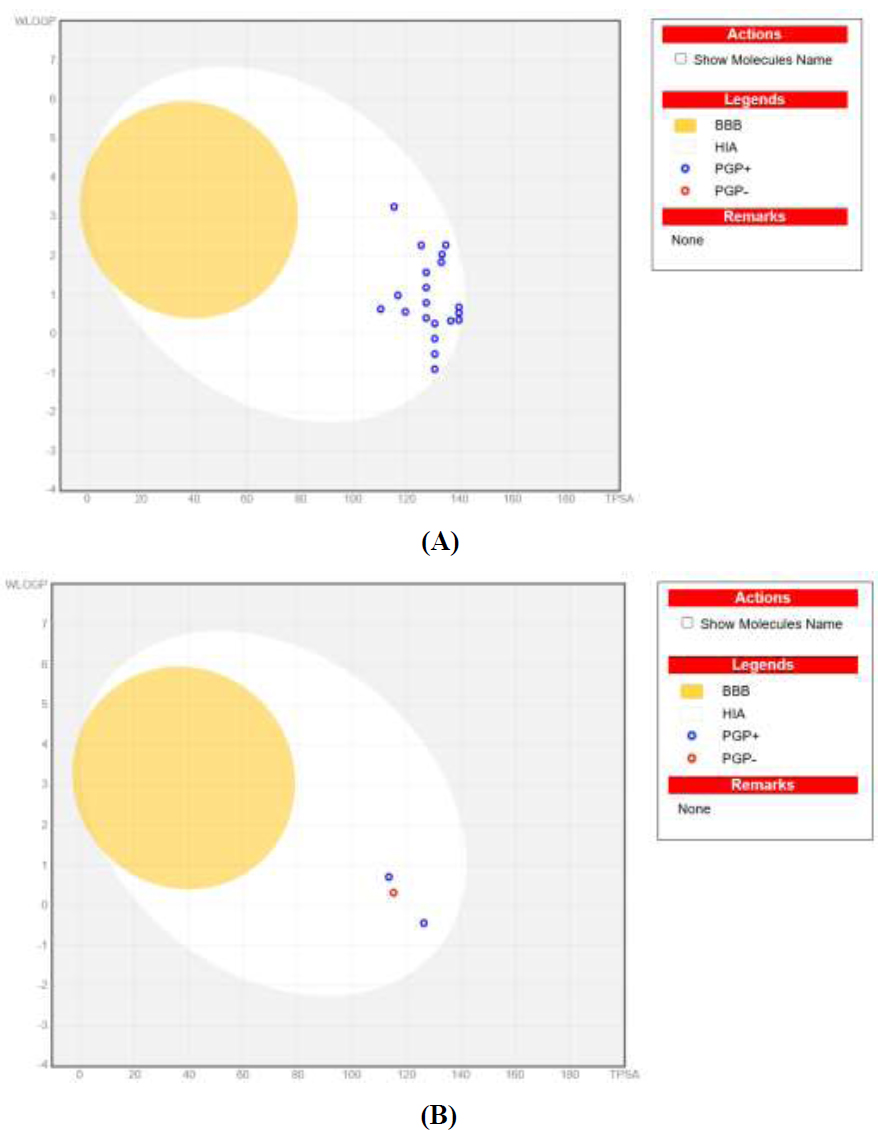
BOILED-Egg predicted models of (A) 20 newly designed and (B) reported compounds 23, 42, and 69.
For CNS drug discovery, overcoming the challenge of blood–brain barrier penetration is critical, as the BBB consists of tightly bound endothelial cells that restrict drug transport [63, 68]. While the proposed compounds did not exhibit BBB penetration, their favorable ADMET properties and adherence to Lipinski’s rule highlight their potential for non-CNS therapeutic applications [69, 70]. These findings demonstrate the utility of SwissADME in guiding the rational design and evaluation of drug candidates, ensuring improved efficacy, safety, and pharmacokinetic profiles [71, 54].
In a metabolism analysis, the enzymatic metabolism of drugs, primarily mediated by the cytochrome P450 (CYP) enzyme family, plays a crucial role in drug biotransformation and detoxification [72, 73]. CYP enzymes, particularly isoforms such as CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4, are responsible for metabolizing more than 90% of drugs during the first phase of metabolism [74-76]. Among these, CYP2D6 and CYP3A4 are the primary enzymes involved in drug metabolism [77, 78].
Swiss ADME was used to evaluate the inhibitory or non-inhibitory behavior of the proposed compounds against major CYP enzymes. Enzyme inhibition is a critical factor in metabolism-based drug–drug interactions [79], as it can impair drug clearance, leading to elevated plasma concentrations and potential toxicity [80, 81]. Conversely, for prodrugs, CYP inhibition can reduce therapeutic efficacy [82, 83]. Importantly, all proposed compounds were predicted to be non-inhibitors of CYP2D6, as shown in Table 8. This finding suggests that the compounds are likely to undergo effective metabolism without causing hepatic dysfunction or adverse drug interactions. By adhering to these metabolic criteria, the compounds exhibit favorable profiles for safe and efficient biotransformation, ensuring their potential for therapeutic development [84].
Additionally, the bioavailability assessment revealed that all the newly developed compounds are predicted to exhibit excellent oral bioavailability in humans. This conclusion is supported by the fact that all tested molecules fall within the pink region of the bioavailability radar shown in Fig. (6) [85, 86]. This region represents the optimal range based on key physicochemical properties, including flexibility, saturation, solubility, lipophilicity, polarity, and molecular size [87, 88]. The bioavailability profile of the designed compound was evaluated in comparison with the predicted bioavailability profile of the most active reported MMP-2 inhibitor, as determined using SwissADME. The analysis confirmed that the designed compound possesses a valid and favorable bioavailability profile.
| Compound | GI | BBB | PGP | C1A2 | C2C19 | C2C9 | C2D6 | C3A4 | log Kp (cm/s) |
|---|---|---|---|---|---|---|---|---|---|
| M1 | High | No | Yes | No | No | No | No | No | -8.72 |
| M2 | High | No | Yes | No | No | No | No | No | -8.66 |
| M3 | High | No | Yes | No | No | No | No | No | -8.48 |
| M4 | High | No | Yes | No | No | No | No | No | -8.26 |
| M5 | High | No | Yes | No | No | No | No | No | -8.21 |
| M6 | High | No | Yes | No | No | No | No | No | -9.05 |
| M7 | High | No | Yes | No | No | No | No | No | -8.87 |
| M8 | High | No | Yes | No | No | No | No | No | -8.65 |
| M9 | High | No | Yes | No | No | No | No | No | -8.61 |
| M10 | High | No | Yes | No | No | No | No | No | -7.86 |
| M11 | High | No | Yes | No | Yes | No | No | No | -7.63 |
| M12 | High | No | Yes | No | No | No | No | No | -7.44 |
| M13 | High | No | Yes | No | No | No | No | No | -8.11 |
| M14 | High | No | Yes | No | No | No | No | No | -8.3 |
| M15 | High | No | Yes | No | No | No | No | No | -8.24 |
| M16 | High | No | Yes | No | No | No | No | No | -7.6 |
| M17 | High | No | Yes | No | No | No | No | No | -8.04 |
| M18 | High | No | Yes | No | No | No | No | No | -9.05 |
| M19 | High | No | Yes | No | No | No | No | No | -8.66 |
| M20 | High | No | Yes | No | No | No | No | No | -8.88 |
| Ref. (23) | High | No | Yes | No | No | No | No | No | -8.84 |
| Ref. (42) | High | No | Yes | No | No | No | No | No | -8.45 |
| Ref. (69) | High | No | No | No | No | No | No | No | -8.25 |
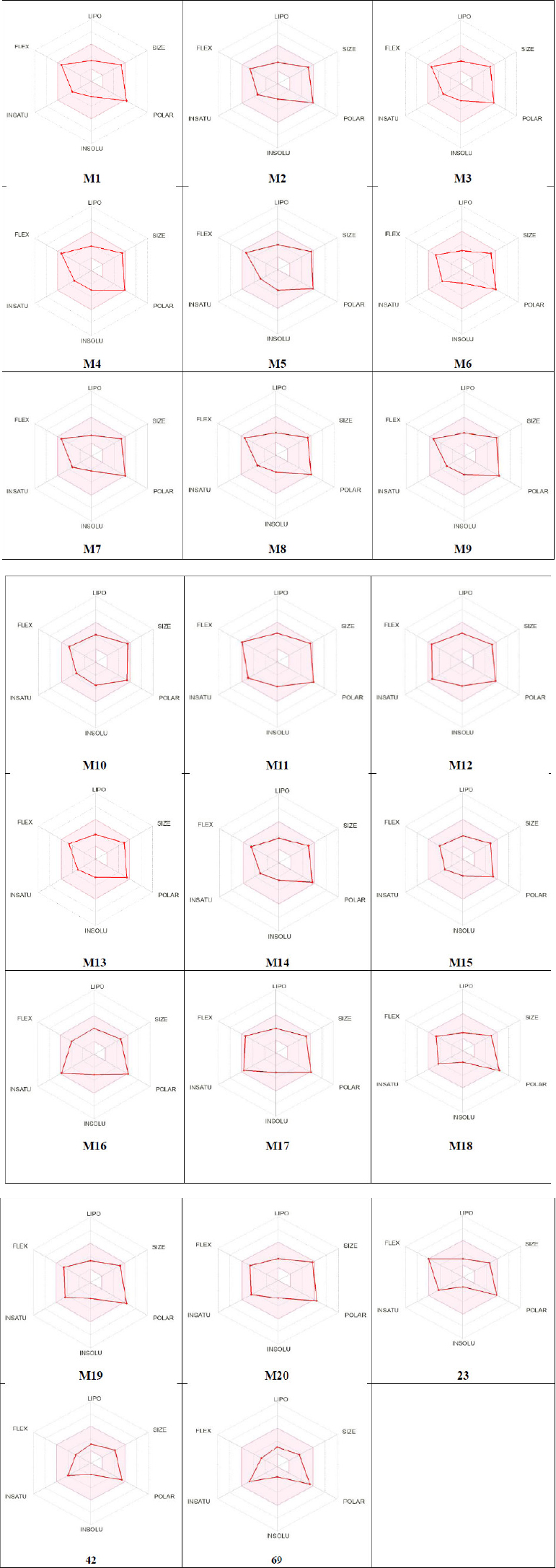
Bioavailability radars for 20 designed molecules (M1-M20) and compounds 23, 42, and 69.
CONCLUSION
This study successfully integrates QSAR modeling, molecular docking, and ADMET predictions to explore the anticancer potential of pyrrolidine derivatives as matrix metalloproteinase-2 (MMP-2) inhibitors. The developed 2D-QSAR model demonstrated robust predictive power with reliable statistical parameters, enabling the design of novel derivatives with enhanced MMP-2 inhibitory activity. Key compounds, including M10 and M17, emerged as promising candidates, exhibiting strong binding affinities, favorable ADME profiles, and improved biological activity predictions.
The findings highlight the significance of calculated molar refractivity, hydrophobicity, and specific substituents such as CONHOH and SO2 p-CH3C6H4 groups in modulating MMP-2 inhibition. Molecular docking studies revealed favorable interactions between the designed compounds and the active site of MMP-2, while ADMET analysis indicated their potential for further development.
This investigation underscores the value of computer-aided drug design (CADD) in reducing costs and streamlining the discovery of potent anticancer agents. The identified compounds, particularly M10 and M17, warrant further synthesis and pharmacological evaluation to optimize their properties and validate their efficacy as MMP-2 inhibitors. These findings provide a strong foundation for advancing pyrrolidine derivatives as targeted therapies for cancer treatment.
AUTHORS’ CONTRIBUTIONS
The authors confirm their contribution to the paper as follows: R.K.Y.: Methodology; P.C., V.M.P.: Draft manuscript. All authors reviewed the results and approved the final version of the manuscript.
LIST OF ABBREVIATIONS
| ECM | = Extracellular matrix |
| MLR | = Multiple linear regression |
| QSAR | = Quantitative Structure-Activity Relationship |
| TIMPs | = Tissue inhibitors of metalloproteinases |
| CMR | = Calculated molar refractivity |
AVAILABILITY OF DATA AND MATERIALS
All data generated or analyzed during this study are included in this published article.
ACKNOWLEDGEMENTS
Declared none.

