All published articles of this journal are available on ScienceDirect.

Design and Synthesis of Imidazopyrimidine Derivatives as Potent iNOS Dimerization Inhibitors
Authors Info & Affiliations
Abstract
A series of imidazopyrimidine derivatives with the general formula I was synthesized and identified as potent inhibitors of iNOS dimer formation, a prerequisite for proper functioning of the enzyme. Stille and Negishi coupling reactions were used as key steps to form the carbon-carbon bond connecting the imidazopyrimidine core to the central cycloalkenyl, cycloalkyl and phenyl ring templates.
Nitric oxide (NO) is involved in many physiological processes, such as maintaining vascular tone and homeostasis, mediating neurotransmission, regulating gastrointestinal motility, and enacting host defense in the immune system [1-3]. In mammals, nitric oxide (NO) is synthesized from L-arginine by a family of NO synthases (NOS). Of the three characterized NOS isoforms, the neuronal NOS (nNOS) and endothelial NOS (eNOS) are constitutively expressed, and under normal physiological conditions, generate low levels of NO in response to increases in intracellular calcium levels. The third NOS isoform, the inducible NOS (iNOS), is calcium-independent, not usually expressed under physiological conditions, and is induced by endotoxin and/or cytokines, such as lipopolysaccharide (LPS), interleukin-1β (IL-1β), tumor necrosis factor α (TNF-α) and interferon-γ (IFNγ). Once induced, iNOS produces high and sustained levels of NO. The overexpression of iNOS, and the resulting excessive production of NO which results in cellular cytotoxicity and tissue damage, has been implicated in the pathogenesis of a number of inflammatory diseases, such as rheumatoid arthritis, osteoarthritis, inflammatory bowel disease, multiple sclerosis and asthma [3-8]. Therefore, iNOS inhibitors may find utility for the treatment of these diseases. Because of the importance of the constitutive forms in normal physiology, high selectivity for iNOS is advantageous to avoid blocking the basic homeostatic functions of the eNOS and nNOS isoforms. The three NOS isoforms differ in their location and function, but are similar in that they are only active in the dimeric form [9-1]. Preventing the dimerization of inactive NOS monomers into active homodimers has emerged as a novel pharmacological strategy to develop isoform-selective NOS inhibitors. Highly potent and selective imidazopyri-midine-based iNOS dimerization inhibitors, exemplified by compounds 1 and 2 (Fig. 1), were discovered recently. These compounds significantly decreased levels of NO production [10, 11]. Based on the crystal structure of 2 bound to murine iNOS monomeric oxygenase domain (iNOS Δ114) [12-14], the imidazole group binds to the heme, while the benzodioxolane group fits closely between residues in the iNOS monomer active site and the pyrimidine ring, resulting in a U-shaped conformation of the molecule in its active site. This prevents Glu377 of helix 7A from occupying the position that leads to dimer formation. Based on this binding mode, new inhibitors using alternative linkers such as hydroxyethylamine, hydroxypiperidine, hydroxypyrimidine, etc, to connect the benzodioxolane and imidazole moieties have been reported [12-14]. As part of our research program on new chemical classes of iNOS inhibitors, we designed and synthesized a series of imidazopyrimidine derivatives with the general formula I (Fig. 1) as isosteric analogs of 1 and 2. In the structure of these compounds, the central piperazine and pyrrolidine heterocycle templates in 1 [10, 11] and 2 [11] were replaced with cycloalkenyl, cycloalkyl and phenyl rings. Some of these new agents were potent iNOS dimerization inhibitors in cell-based iNOS assays.
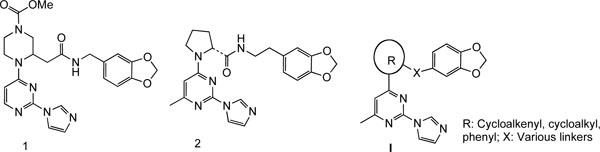
In compounds 1 and 2, the piperazine and pyrrolidine heterocycles are connected to the pyrimidine ring via a carbon-nitrogen (C-N) bond which can be easily formed by a simple nucleophilic substitution reaction [14]. In our proposed target molecules, the pyrimidine ring is linked to a cycloalkenyl, cycloalkyl or phenyl ring via a carbon-carbon (C-C) bond, which was expected to be established through palladium-catalyzed coupling reactions. The synthesis of the target molecules 3-9 is outlined in Scheme 1. We first attempted to prepare these compounds using the palladium-catalyzed Suzuki coupling as the key step. Condensation of the commercially available 2,4-dichloro-6-methylpyrimidine (17) with imidazole in acetone in the presence of potassium carbonate gave a mixture of regioisomers 18 and 19 easily separated by column chromatography [14]. The enol triflates 21a,b were prepared from the corresponding ketoesters 20a,b as reported previously [15]. Palladium catalyzed reaction of the enol triflate 21a with bis(pinacolato)diboron [16] was incomplete after 24 hours at 50 oC, and furthermore, the desired boron derivative 22 could not be separated from the bis(pinacolato)diboron by column chromatography. Prolonging the reaction time resulted in a mixture of 22 and the dimer resulting from the coupling of 22 with the triflate 21a. The inseparable mixture 22/bis(pinacolato)diboron was then reacted with the chloropyrimidine derivative 19 under Suzuki coupling conditions. Unfortunately, the reaction produced a complex mixture, failing to yield the desired coupling product. An alternative route for the preparation of 24a was then designed. This involved first converting 19 to the corresponding pinacolatoboron intermediate, and then coupling it with the triflates 21a under Suzuki coupling conditions. However, condensation of 19 with bis(pinacolato)diboron using the same reaction condition as for 22 was not successful with no reaction observed. With the failure of the Suzuki coupling approach, next we tried other alternatives. Palladium-catalyzed reductive coupling reaction of chloropyrimidine 19 with methyl 1-cyclopentene-1-carboxylate using the same reaction condition as described in a previous total synthesis of epibatidine [17], afforded the deschloro compound, i.e. 2-(1H-imidazol-1-yl)-4-methylpyrimidine, instead of the desired coupled product 24a. Heck reaction of chloropyrimidine 19 with methyl 1-cyclopentene-1-carboxylate following Fu’s modified method using Pd2(dba)3-P(t-Bu)3 as catalyst [18] gave no reaction. Another alternative approach to secure the C-C bond connecting the cycloalkenyl moiety to the pyrimidine core is through the Stille coupling method. Treatment of 19 with bis(tributyltin) using Pd(Ph3P)2Cl2 as catalyst in DMF furnished the desired tin compound 23 in 74% yield. Condensation of 23 with the enol triflate derivative 21a under standard Stille coupling reaction condition using Pd(PPh3)4 as catalyst in the presence of LiCl in refluxing THF [19] gave no reaction. Changing the solvent from THF to dioxane did not result in any improvement. Coupling of 23 with the triflate 21a also failed with Corey’s modified coupling system using Pd(PPh3)4/CuCl/LiCl [20]. Finally we found that when the Stille coupling reaction of 23 with the triflate 21a was carried out in DMF at 110 oC using Pd(PPh3)2Cl2 as catalyst, the desired coupling product 24a was successfully obtained, although in low yield (~ 10%). Replacing Pd(PPh3)2Cl2 with dichlorobis(tri-o-tolylphosphine)palladium (II), i.e. Pd[(o-CH3Ph)3P)2]Cl2, moderately improved the yield to 16%. To our delight, while using dichlorobis(acetonitrile) palladium (II), Pd(CH3CN)2Cl2, which is not a commonly used catalyst for Stille coupling, the coupling product 24a was obtained in 38% isolated yield. This catalyst also worked efficiently for the coupling of the enol triflate 21b with 23, yielding 24b in similar yield. The coupling system we describe here, 0.1 equiv Pd(CH3CN)2Cl2 / 6 equiv LiCl / DMF, may have potential applications in other difficult cases of Stille coupling reactions. With the success of introducing the five-membered cyclopentenyl and the six-membered cyclohexenyl groups to the pyrimidine ring via C-C bond, the following conversion of 24a,b to the target molecules was straightforward. Hydrolysis of the esters 24a,b with lithium hydroxide afforded the carboxylic acids 25a,b, which were coupled with 3,4-methylenedioxyphen-ethylamine using TBTU [O-(benzotriazol-1-yl)-N,N,N',N'-tetra-methyluronium tetrafluoroborate] as coupling reagent to yield the target molecules 3 and 6, respectively. Hydrogenation of 3 and 6 gave the double bond reduced cis analogs 4 and 7, respectively. Compounds 4 and 7 were cleanly epimerized to the trans analogs 5 and 8 by treatment with DBU in refluxing benzene. The synthesis of the target compound 9 was also straightforward. The reaction of chloropyrimidine 19 with 2-ethoxycarbonylphenylzinc bromide in the presence of Pd(PPh3)4 under Negishi coupling condition afforded the coupled product 26 in 84% yield. The ester 26 was then converted to the target compound 9 in a similar manner as for the synthesis of 3 and 6 from 24a,b.
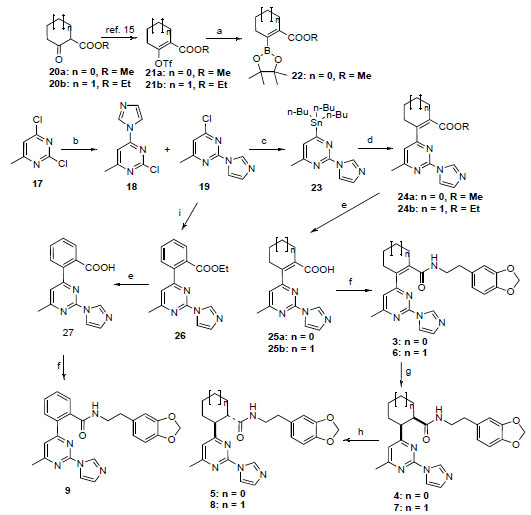
Reagents and conditions: a) bis(pinacolato)diboron, K2CO3, Pd(PPh3)2Cl2, PPh3, dioxane, 50 °C, b) imidazole, K2CO3, acetone, rt, 37% for 19; c) bis(tributyltin), Pd(PPh3)2Cl2, DMF, 95 °C, 24 h, 74%; d) 21a, b, Pd(CH3CN)2Cl2, LiCl, DMF, 105 °C, 72 h, 38% for 24a, 36% for 24b; e) LiOH, MeOH-THF-H2O, rt, 18 h, 92% for 25a, 88% for 25b, 84% for 27; f) 3,4-methylenedioxyphenethylamine, TBTU, iPr2NEt, CH3CN, rt, 48 h, 88% for 3, 92% for 6, 70% for 9; g) H2 (1 atm), Pd/C, rt, 72 h, > 90%; h) DBU, PhH, reflux, 48 h, 90%; i) 2-ethoxycarbonyl-phenylzinc bromide, Pd(PPh3)4, THF, 50 °C, 48 h, 87%.
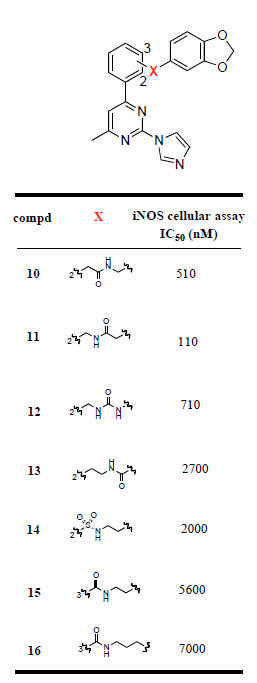
Next, we made various modifications on the molecule 9 at the tether linking the middle phenyl ring to the benzodioxolane group to further investigate the SAR of this new chemical series. The compounds 10-16 were prepared according to Scheme 2. 2-Iodophenylacetic acid (28) was condensed with piperonylamine using TBTU as coupling reagent to provide the amide 29, which was then coupled with the organotin derivative 23 using Pd(CH3CN)2Cl2 as catalyst under microwave conditions to yield 10. Similar to the preparation of ester 26, Negishi coupling of 19 with 2-cyanophenylzinc bromide furnished 30 in excellent yield. The cyano derivative 30 was then converted to the primary amine 31 by hydrogenation. Compound 31 was then converted to the amide 11 using the above-mentioned TBTU coupling method, and converted to the urea analog 12 by condensation with 3,4-(methylenedioxy) phenyl isocyanate. Stille coupling of bromide 33 and 35 with 23 using the same reaction condition as for 24a,b yielded amide 13, and sulfonamide 14, respectively. Compounds 15 and 16 were prepared according to the same reaction conditions described above for the synthesis of 9.
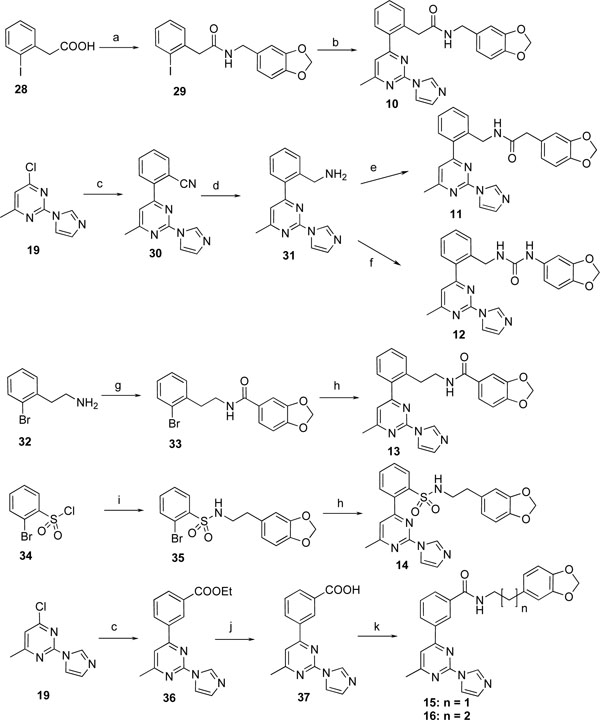
Reagents and conditions: a) piperonylamine, TBTU, iPr2NEt, CH3CN, rt, 24 h, 98%; b) 23, Pd(CH3CN)2Cl2, LiCl, Et3N, DMF, 180 °C, microwave, 1 h, 38%; c) 2-cyanophenylzinc bromide or 3-ethoxycarbonylphenylzinc iodide, Pd(PPh3)4, THF, 50 °C, 48 h, 94% for 30, 49% for 36; d) H2 (1 atm), Pd/C, MeOH, HCl, rt, 48 h, 85%; e) 3,4-(methylenedioxy)phenylacetic acid, TBTU, iPr2NEt, CH3CN, rt, 24 h, 93%; f) 3,4-(methylenedioxy)phenyl isocyanate, Et3N, DCM, rt, 2 h, 91%; g) piperonyloyl chloride, Et3N, DCM, rt, 2 h, 100%; h) 23, Pd(CH3CN)2Cl2, LiCl, DMF, 105 °C, 72 h, 4% for 13, 10% for 14; i) 3,4-methylenedioxyphenethylamine, Et3N, DCM, rt, 1 h, 100%; j) LiOH, MeOH-THF-H2O, rt, 24 h, 86%; k) 3,4-methylenedioxyphenethylamine or 3-benzo[1,3]dioxol-5-yl-propylamine, TBTU, iPr2NEt, CH3CN, rt, 48 h, 83% for 15, 20% for 16.
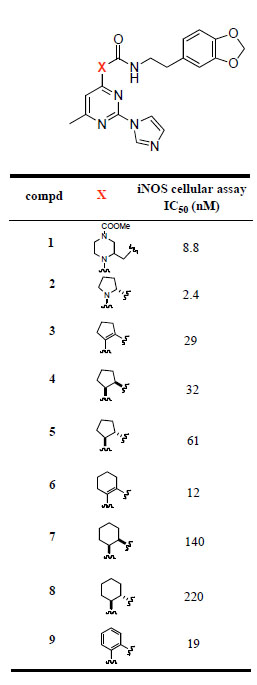
The imidazopyrimidines 3-16 were evaluated for their abilities to inhibit cytokine-mediated induction of iNOS activity in DLD-1 cells (Tables 1 and 2). The initial strategy consisted of replacing the central pyrrolidine group of 2 with a cyclopentene moiety. This modification led to a 10-fold decrease in the iNOS potency. Hydrogenation of the double bond functionality of 3 provided compound 4, which displayed similar iNOS potency than its unsaturated analog. The cis and trans relative stereochemistry at the cyclopentyl scaffold did not significantly influence the activity, the iNOS potency of the the cis and trans isomers 4 and 5 was within a factor of 2. Extending the cyclopentene group of 3 to a cyclohexene moiety led to a 2-fold increase in the iNOS potency. However, the cis and trans saturated analogs of 6, i.e. compounds 7 and 8 respectively, were more than 10-fold less potent than 6. Replacement of the cyclohexenyl group of 6 with a phenyl ring was well tolerated. The phenylpyrimidine derivative 9 had comparable iNOS inhibitory activity as its cyclohexenyl analog (6: IC50 = 12 nM; 9: IC50 = 19 nM). In order to explore the size of the pocket in which the benzodioxolane group of 9 interacts, we prepared various analogs of 9 in which the benzodioxolane moiety is connected to the 2-(1H-imidazol-1-yl)-4-methyl-6-phenyl-pyrimidine template by various linkers. As indicated in Table 2, replacement of the carbonylaminoethyl linker (CONHCH2CH2) of 9 with various chains containing either amido (compounds 10, 11, 13) or urea (compound 12) functionalities led to a significant decrease in the iNOS potency. Similarly, replacing the carboxamide functionality of 9 by a sulfonamide group (compound 14) led to a 100-fold decrease in the iNOS potency. Furthermore, changing the ortho-substitution pattern of the central phenyl ring in 9 to the meta-substitution pattern as in 15 led to a dramatic loss of potency. Taken together, these data indicate that the structure and the connection pattern of the tether linking the central phenyl ring of the 2-(1H-imidazol-1-yl)-4-methyl-6-phenylpyrimidine to the benzodioxolane group had a significant influence on the iNOS potency. The four atom amide tether, CONHCH2CH2, connecting to the central phenyl ring with an ortho-substitution pattern was the most preferred for optimal iNOS inhibitory activity. Modifications on this linker were generally detrimental to the potency.
iNOS Inhibitory Activity of Compounds 1-9
 |
iNOS Inhibitory Activity of Compounds 10-16
 |
In summary, a series of imidazopyrimidine derivatives 3-16 was synthesized and evaluated to inhibit cytokine-mediated induction of iNOS activity in DLD-1 cells. Stille coupling and Negishi coupling reactions were used as key steps to form the C-C bond connection between the pyrimidine and the central cycloalkenyl, cycloalkyl and phenyl rings. Some of these agents are potent inhibitors of iNOS dimer formation, a prerequisite for proper functioning of the enzyme in cell-based iNOS assays. Among these, compounds 6 and 9, which contain six-membered cyclo-hexenyl and phenyl rings as central templates, are the most potent inhibitors in the new chemical series, displaying IC50 values of 12 and 19 nM, respectively. These compounds may serve as good leads for further analog design, and the synthetic methods can provide a general approach to the preparation of such compounds.

