All published articles of this journal are available on ScienceDirect.

Metabotropic Glutamate Receptors: Potential Drug Targets for Psychiatric Disorders
Abstract
Metabotropic glutamate receptors (mGlu receptors) have emerged as new therapeutic targets for psychiatric disorders, such as schizophrenia, depression and anxiety with their regulatory roles in glutamatergic transmissions. To date, several ligands selective for each mGlu receptor have been synthesized, and pharmacological significances of these ligands have been demonstrated in animal models. Among them, mGlu2/3 receptor agonists have been proven to be effective for treating schizophrenia and anxiety disorders in clinical studies, which may prove utilities of mGlu receptor ligands for the treatment of psychiatric disorders. This article reviews recent advances in development of each mGlu receptor ligands and their therapeutic potential.
1. INTRODUCTION
Glutamate is the major excitatory neurotransmitter in the brain. It is involved in a wide range of physiological processes in the central nervous system that are associated with emotion, cognition and motor functions. Glutamate receptors are categorized into 2 major types: ionotropic glutamate receptors (iGlu receptors), in which the receptors have an ion channel structure; and metabotropic glutamate receptors (mGlu receptors), which are coupled to G-proteins [1, 2]. iGlu receptors, which facilitate fast synaptic transmission, are also classified into N-methyl-D-aspartate (NMDA), α-amino-3-hydroxy-5-methyl-isoxazole-4-propionate (AMPA) and kainate receptors [1, 2]. mGlu receptors also have several isoforms (mGlu1–mGlu8).
mGlu receptors are currently classified into 3 groups based on their sequence homology, second messenger coupling, and pharmacological characteristics (Table 1) [3-5]. Group I mGlu receptors include mGlu1 and mGlu5 that are coupled to phospholipase C, while both group II mGlu receptors (mGlu2 and mGlu3) and group III mGlu receptors (mGlu4, mGlu6, mGlu7, and mGlu8) are negatively coupled to adenylyl cyclase activity. Among these mGlu receptors, numerous ligands (agonists, antagonists, positive modulators or negative modulators) have been developed for the mGlu2/3 and mGlu5 receptors. Studies using these ligands have been established their physiological and pharmaco-logical significance as well as their possible therapeutic applications.
Classification and Characteristics of mGlu Receptors
| Group I | Group II | Group III | ||||||
|---|---|---|---|---|---|---|---|---|
| mGlu1 | mGlu5 | mGlu2 | mGlu3 | mGlu4 | mGlu6 | mGlu7 | mGlu8 | |
| Structure | Class C GPCR | Class C GPCR | Class C GPCR | |||||
| Signaling | Gq/11 Activation of phospholipase C | Gi/o Inhibition of adenylyl cyclase | Gi/o Inhibition of adenylyl cyclase | |||||
| Agonists | DHPG | CHPG | LY404039, LY354740, MGS0028, MGS0008 | LY404039, LY354740, MGS0028, MGS0008 | L-AP4, PHCCC | HomoAMPA | L-AP4, AMN082 | L-AP4, RS-PPG |
| Positive allosteric modulators | DFB, CDPPB, CPPHA, ADX47273 | LY487379, BINA | AMN082 | |||||
| Antagonists | JNJ16567083, LY367385 | MPEP, MTEP | LY341495, MGS0039 | LY341495, MGS0039 | CPPG | CPPG | CPPG | CPPG |
A growing body of evidence shows that manipulations of the mGlu2/3 and mGlu5 receptors exhibit significant pharmacological effects in numerous animal models, including models for psychiatric disorders. In addition, mGlu2/3 receptor agonists have demonstrated efficacy in clinical studies in patients with schizophrenia [6] or anxiety disorders [7-9]. This review will focus on recent developments of agonists/antagonists, including allosteric modulators for mGlu receptors and the possible therapeutic application of these ligands for the treatment of psychiatric disorders, such as schizophrenia and depression.
2. SCHIZOPHRENIA
2.1. Role of Glutamatergic Transmission in Schizophrenia
In addition to the well-established “dopamine hypothesis of schizophrenia,” it has also been suggested that abnormalities of glutamate transmission are involved in the pathophysiology of schizophrenia. Significantly lower levels of glutamate are found in the cerebrospinal fluid (CSF) and in postmortem brain tissues of schizophrenic patients [10, 11]. CSF glutamate levels are also inversely correlated with the severity of positive symptoms in unmedicated patients [12]. Phencyclidine (PCP) and ketamine, non-competitive NMDA receptor antagonists, produce transient psychoses, disrupted affect and caused cognitive impairments in healthy volunteers, all of which are similar to what are observed in schizophrenia [13-16], and lead to profound exacerbations of pre-existing symptoms in schizophrenic patients [15]. Moreover, NMDA receptor antagonists and transgenic manipulation of an NMDA receptor subunit (NR1) induce schizophrenia-like behavioral abnormalities (increased locomotor activity, reduced social interaction, cognitive dysfunction), which are ameliorated by antipsychotic treatments [17-19].
Thus, glutamatergic dysfunction, and hypofunction of NMDA receptors in particular, may have an important role in the pathophysiology of schizophrenia. Because mGlu receptors have modulatory roles on NMDA receptor-mediated transmission, mGlu receptor ligands may be attractive therapeutic targets for the treatment of schizophrenia.
2.2. mGlu2/3 Receptor Agonists/mGlu2 Receptor Potentiators
In recent years, several lines of evidence have shown that mGlu2/3 receptor agonists may be effective for the treatment of schizophrenia in both animal models and in humans. mGlu2/3 receptor agonists, such as (1S,2S,5R,6S)-2-aminobicyclo[3.1.0]hexane-2,6-dicarboxylic acid (LY3547 40), have been reported to attenuate locomotor hyperactivity induced by an NMDA receptor antagonist in rodents [20]. Similar effects were observed with other mGlu2/3 receptor agonists, including (-)-(1R,4S,5S,6S)-4-amino-2-sulfony-lbicyclo[3.1.0]hexane-4,6-dicarboxylic acid (LY404039) [21], (1S,2R,5R,6R)-2-amino-4-oxabicyclo[3.1.0]hexane-2,6-dicarboxylic acid (LY379268) [22], (1S,2S,3S,5R,6S)-2-amino-3-fluorobicyclo[3.1.0]hexane-2,6-dicarboxylic acid (MGS0008), and (1R,2S,5S,6S)-2-amino-6-fluoro-4-oxobicyclo[3.1.0]hexane-2,6-dicarboxylic acid (MGS0028) [23]. Moreover, mGlu2/3 receptor agonists have been shown to inhibit amphetamine-induced locomotor hyperactivity [21, 24] and conditioned avoidance responding [21, 25]. These models may be predictive of the efficacy for positive symptoms of schizophrenia. Therefore, mGlu2/3 receptor agonists may be effective for the treatment of positive symptoms of schizophrenia.
The effect of LY404039 on PCP- or amphetamine-induced locomotor hyperactivity is absent in mGlu2 receptor- or mGlu2/3 receptor-null mice, but not in mGlu3 receptor-null mice [26]. In addition, the effects of mGlu2/3 receptor agonists on PCP- or amphetamine-induced locomotor hyperactivity were mimicked by the selective mGlu2 receptor potentiator N-(4-(2-methoxyphenoxy)phenyl)-N-(2,2,2-trifluoroethylsulfonyl)pyrid-3-ylmethylamine (LY487379) [24]. Thus, the mGlu2 receptor, but not the mGlu3 receptor, is responsible for the antipsychotic actions of mGlu2/3 receptor agonists.
LY354740 improved PCP-impaired performance in a T-maze discrete-trial delayed alternation task [20] and reversed the deficits of social discrimination induced by neonatal treatment with PCP [27]. These findings suggested the potential of mGlu2/3 receptor agonists to improve the cognitive dysfunctions observed in schizophrenic patients. However, there are some contradictory findings indicating that mGlu2/3 receptor agonists may not improve cognitive dysfunction, and may even cause it to deteriorate.
For example, LY354740 induced working memory deficits in delayed matching and non-matching to position and spatial memory tasks in a Morris water maze [28], and did not improve PCP-induced cognitive deficits in a spontaneous alternation task and passive avoidance task [29]. Moreover, mGlu2/3 receptor agonists did not reverse amphetamine- or PCP-induced disruption of prepulse inhibition (PPI) [24, 30], a fundamental form of information processing that is impaired in schizophrenic patients.
In contrast, a selective mGlu2 receptor potentiator, LY487379, significantly improved amphetamine-disrupted PPI [24], and also significantly improved social discrimination deficits induced after neonatal treatment with PCP [28]. Based on these findings, the effects of mGlu2/3 receptor agonists may be task-dependent or influenced by the conditions that were used. It is also conceivable that selective mGlu2 receptor stimulation may be important in order to fully exert its effects on cognitive dysfunction, while mGlu3 receptor stimulation may have opposite effects.
The neuronal mechanisms by which mGlu2/3 receptor agonists exert their effects have been investigated. mGlu2/3 receptor agonists are considered to exert their effects by acting on the glutamatergic, dopaminergic, and 5-HTergic pathways. Administration of NMDA receptor antagonists increases glutamate release in the prefrontal cortex, and also elicits working memory dysfunction [20, 31, 32]. Thus, it is presumed that NMDA receptor dysfunction in the prefrontal cortex increases glutamate release and, thereby, impairs working memory.
mGlu2/3 receptor agonists have been shown to attenuate the increased glutamate release caused by blockade of NMDA receptors. Consistent with this result, a marked increase in relative cerebral blood volume, which reflects hypermetabolism associated with increased glutamate release, was seen in the prefrontal cortex in rats following administration of PCP, as determined by pharmacological magnetic resonance imaging. LY354740 attenuated this metabolic hyperactivity [33]. Given that prefrontal abnormallities are assumed to be involved in the etiology of schizophrenic symptoms, attenuation of prefrontal abnormalllities (glutamate overflow and hypermetabolism) may contribute to the antipsychotic actions of mGlu2/3 receptor agonists.
Moreover, an mGlu2/3 receptor agonist inhibited PCP-increased dopamine flow in the nucleus accumbens shell [34], while it increased dopamine release in the prefrontal cortex [21, 35]. Given that accumbal dopaminergic hyperactivity is associated with positive symptoms of schizophrenia, while prefrontal dopaminergic hypoactivity is associated with impairment of cognition, these differential effects of mGlu2/3 receptor agonists could contribute to the antipsychotic effects of mGlu2/3 receptor agonists.
Recently, interactions between the 5-HT2A and mGlu2 receptors have been suggested. LY354740 attenuated serotonin-induced excitatory postsynaptic currents recorded in layer V pyramidal cells of the medial prefrontal cortex, suggesting that mGlu2/3 receptors negatively regulated 5-HT2A receptor-mediated responses to stimulate glutamate release, presumably from thalamocortical afferents to the prefrontal cortex. Moreover, LY354740 attenuated 5-HT2A receptor agonist-induced head shakes in rats [36]. Attenuation of 5-HT2A receptor-mediated electrophysio-logical and behavioral responses was mimicked by an mGlu2 receptor potentiator [37], indicating that the mGlu2 receptor, but not the mGlu3 receptor, was involved in this event. It was also recently shown that the 5-HT2A and mGlu2 receptors formed a heterodimeric complex [38]. Thus, it is intriguing to assume that the mGlu2 receptor may exert its effects, in part, by inhibiting 5-HT2A receptor signaling via the mGlu2 receptor-5-HT2A receptor complex.
The efficacy of an mGlu2/3 receptor agonist, LY2140023 (a methionine amide of LY404039), was reported in a randomized, double-blinded, placebo-controlled study of 196 schizophrenic patients [6]. LY2140023 significantly improved both positive and negative symptoms, as observed with olanzapine. LY2140023 neither increased prolactine nor worsened extrapyramidal symptoms. Furthermore, LY2140 023 did not affect body weight unlike olanzapine, which causes increased body weight. Therefore, mGlu2/3 receptor agonists may be effective and safe as monotherapy for the treatment of schizophrenia, although larger and longer-term studies are needed to verify this.
2.3. mGlu5 Receptor Agonists/mGlu5 Receptor Potentiators
(RS)-2-Chloro-5-hydroxyphenylglycine (CHPG), an mGlu5 receptor agonist, has been reported to attenuate ketamine-induced increases in locomotor hyperactivity, and to improve PPI disruption and object recognition memory impairment induced by ketamine [39]. Similarly, CHPG ameliorated PPI disruption induced by amphetamine [40], indicating that mGlu5 receptor stimulation exerts antipsychotic activity in animal models. mGlu5 receptor potentiators offer an attractive alternative to the direct mGlu5 receptor activation by orthosteric competitive agonists.
To date, several structurally diverse mGlu5 receptor potentiators have been reported, including 3,3’-difluoro-benaldazine (DFB), S-(4-fluoro-phenyl)-{3-[3-(4-fluoro-phenyl)-[1,2,4] oxadiazol-5-yl]-piperidin-1-yl}-methanone (ADX47273) and 3-cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide (CDPPB). These compounds reportedly prevented locomotor hyperactivity induced by PCP, ketamine, amphetamine, and apomorphine [39, 41, 42]. ADX47273 also reduced conditioned avoidance responding in rats [42]. These findings suggest that mGlu5 receptor potentiation may be effective for the positive symptoms of schizophrenia.
The efficacy of mGlu5 receptor potentiators for cognitive dysfunctions has also been reported. CDPPB improved PPI disruption induced by amphetamine [41]. In a novel object recognition test, CDPPB or DFB improved the impaired recognition capability that was induced by MK-801 or ketamine [42, 43]. CDPPB also attenuated MK-801-impaired task performance in a set-shifting paradigm [44]. Therefore, enhancement of mGlu5 receptor activity with mGlu5 receptor potentiators may be effective for treating the dysfunctions of several cognitive domains associated with schizophrenia.
In addition to improving cognitive deficits, mGlu5 receptor potentiators, including ADX47273, DFB, and CDPPB, also enhanced object recognition memory in a novel object recognition test [42] and spatial memory in Y-maze spatial alternation [45] and Morris water maze tasks [46], indicating that mGlu5 receptor potentiators may have memory-enhancing effects, even for normal conditions. The involvement of the mGlu5 receptor on cognition is supported by a study of mGlu5 receptor-null mice. These mice exhibited reduced long-term potentiation in the CA1 region and dentate gyrus of the hippocampus (NMDA receptor-dependent pathways), but not in the CA3 region (an NMDA receptor-independent pathway). mGlu5 receptor-null mice also displayed impaired spatial learning in a Morris water maze task, impaired contextual learning during fear-condi-tioning [47] and disrupted PPI [40].
The neuronal mechanisms by which mGlu5 receptor potentiation exerts antipsychotic and cognitive enhancing effects have been considered in relation to stimulation of NMDA receptor function. The mGlu5 receptor positively regulates NMDA receptor function. mGlu5 receptor agonists potentiated NMDA receptor function in multiple neuron populations [48, 49], and this was not observed in knockout mice lacking mGlu5 receptors. Similarly, an mGlu5 receptor potentiator, N-{4-chloro-2-[(1,3-dioxo-1,3-dihydro-2H-isoi-ndol-2-yl)methyl]phenyl}-2-hydroxybenzamide (CPPHA), potentiated NMDA currents in hippocampal slices induced by a threshold level of DHPG [50]. In addition, an mGlu5 receptor potentiator, CDPPB, prevented MK-801-induced excessive firing and reduced spontaneous bursting in the medial prefrontal cortex [51]. These findings indicate that mGlu5 receptor stimulation may ameliorate abnormalities in prefrontal cortical neuronal activity, which may be responsible for frontal cortex dependent cognitive function.
Conversely, mGlu5 receptor antagonists potentiated the spontaneous burst and spike activity of cortical neurons [52] and psychotomimetic effects, both of which were induced by NMDA receptor antagonists [49]. Interactions between the mGlu5 and NMDA receptors at the molecular level have been indicated by reports showing that an mGlu5 receptor agonist, DHPG, increased phosphorylation of NMDA NR1 at Ser-897 (a substrate for protein kinase C) in brain slices [53], and that administration of DHPG also increased phosphorylation of NR1 at Ser-897 [54]. Because decreased Ser-897 on the NR1 receptor has been reported in the brains of schizophrenic patients, and antipsychotic drugs increased Ser-897 on NR1 [55, 56], an increase of Ser-897 phosphorylation on NR1 by mGlu5 receptor stimulation is of interest in light of the mGlu5 receptor involvement in schizophrenia.
2. 4. mGlu1 Receptor Antagonists
mGlu1 receptor antagonists such as [4-[1-(2-fluoropyridine-3-yl)-5-methyl-1H-1,2,3-triazol-4-yl]-N-isopropyl-N-methyl-3,6-dihydropyridine-1(2H)-carboxamide] (FTIDC) and 2-cyclopropyl-5-[1-(2-fluoro-3-pyridinyl)-5-methyl-1H-1,2,3-triazol-4-yl]-2,3-dihydro-1H-isoindol-1-one (CFMTI) have been reported to exhibit antipsychotic effects in rodent models of schizophrenia. Of note, these mGlu1 receptor antagonists reduced maximal responses of L-glutamate [57, 58], suggesting that both compounds acted as non-competitive antagonists for the mGlu1 receptor. Thus, FITDC antagonized methamphetamine-induced locomotor hyperactivity and methamphetamine-disrupted PPI [59].
Antipsychotic-like potentials of mGlu1 receptor antagonists have been further characterized by studies using a potent and selective mGlu1 receptor antagonist CFMTI [58]. As observed with FTIDC, at the doses which antagonizes mGlu1 receptor-mediated behavior (face-washing behavior), CFMTI reduced methamphetamine-induced locomotor hyperactivity and disruption of PPI without affecting spontaneous locomotor activity. Moreover, CFMTI attenuated locomotor hyperactivity and PPI disruption induced by ketamine, and reversed MK-801-reduced social interaction (regarded as social withdrawal, one of the negative symptoms). Therefore, based on animal studies, mGlu1 receptor antagonists may have efficacy for positive and negative symptoms, and a certain domain of cognitive dysfunction of schizophrenia.
In contrast, mGlu1 receptor antagonists neither induced catalepsy nor impaired rotarod performance [58, 59], suggesting that mGlu1 receptor antagonists may not cause motor dysfunction, unlike the actions of typical antipsychotics. This atypical activity of mGlu1 receptor antagonists is further supported by c-fos induction, in that CFTI induces c-fos in the medial prefrontal cortex and nucleus accumbens, but not in the dorsolateral striatum, which is in line with the results using clozapine [58].
The neurochemical mechanisms underlying the antipsychotic actions of mGlu1 receptor antagonists are not fully understood. Injection of DHPG, a group I mGlu receptor agonist, into the medial prefrontal cortex increased glutamate release, and an mGlu1 receptor antagonist blocked this [60]. Thus, regulation of glutamate overflow in the medial prefrontal cortex by mGlu1 receptor blockade may be responsible for the pharmacological actions of mGlu1 receptor antagonists.
3. MOOD DISORDERS
3. 1. Role of Glutamatergic Transmission in Depression
Emerging evidence suggests that abnormalities of glutamatergic systems are involved in depression, although the extent and direction of these changes remain subjects of debate. Blood glutamate levels have been shown to increase in medicated patients [61, 62]. Mitani et al. [63] reported that increased glutamate plasma levels reflected the severity of depression. Sanacora et al. [64] reported significantly increased glutamate levels in the occipital cortex in depressed patients as compared to healthy controls.
In contrast, glutamate levels in the anterior cingulate cortex and frontal cortex of patients with major depression were reduced [65, 66], while total REM sleep suppression, an efficient method for relieving depression, produced robust increases in glutamate levels in the pons [67]. Pfleiderer et al. [68] reported reduced Glx (glutamate/glutamine) levels in the left anterior cingulate cortex in severely depressed unipolar patients, which were normalized by electrocon-vulsive therapy. Abnormalities in expression levels of the NMDA receptor and glial type glutamate transporters (EAAT1 and EAAT2) have been reported in postmortem studies of human suicide victims and major depression [69, 70].
In addition, ketamine (an NMDA receptor antagonist) and riluzole (a glutamate modulator) exerted antidepressant activities in patients with major depressive disorders who were unresponsive to conventional antidepressants [71 – 73]. In particular, ketamine reportedly exerted a rapid, sustained and robust antidepressant effect. Thus, modulation of glutamatergic transmission might be an effective approach to treating patients with depression, in particular patients with treatment-resistant depression.
3. 2. mGlu2/3 Receptor Antagonists
Recently, the mGlu2/3 receptor was shown to be involved in depression using selective mGlu2/3 receptor antagonists in rodent models. mGlu2/3 receptor antagonists, MGS0039 and LY341495, shortened immobility times in 2 behavioral despair models, a rat forced swim test, and a mouse tail suspension test [74]. Similar antidepressant effects of an mGlu2/3 receptor antagonist in a despair model were confirmed by Witkin et al. [75]. An antidepressant-like phenotype (reduced immobility time) in a forced swim test was observed in knockout mice lacking mGlu2 receptors [76], suggesting that the mGlu2 receptor may be involved in the antidepressant effects of mGlu2/3 receptor antagonists. In addition, LY341495 attenuated the threshold elevations observed in rats undergoing spontaneous nicotine withdrawal [77]. The threshold elevation following withdrawal from chronic treatment with drugs of abuse has been proposed to reflect anhedonia (loss of interest or pleasure), which is a core symptom of depression [78], and provides additional evidence that mGlu2/3 receptor antagonists may have antidepressant effects. Moreover, subchronic administration of MGS0039 reduced escape failures in a learned helplessness paradigm, indicating antidepressant activity [79].
Neuronal mechanisms underlying the antidepressant effects of mGlu2/3 receptor antagonists have also been investigated. For example, MGS0039 increased the firing rate of the dorsal raphe nucleus and increased serotonin release in the medial prefrontal cortex [80]. This was consistent with the behavioral profile in the forced swim test that showed that mGlu2/3 receptor antagonists selectively increased swimming behavior, as did SSRIs. In addition to serotonergic transmission, MGS0039 increased dopamine release in the nucleus accumbens shell [81], which may explain the anti-anhedonic effect of mGlu2/3 receptor antagonists, as dopaminergic activity in the nucleus accumbens is closely related to reward activity. Also, subchronic administration of MGS0039 increased hippocampal neurogenesis [82], which has been shown to be one mechanism for the actions of antidepressants.
Interestingly, some of the actions of MGS0039 were attenuated by an AMPA receptor antagonist, NBQX [83, 84], suggesting that MGS0039 exerts its effects through AMPA receptor activation. Given that AMPA receptor potentiators have been shown to exhibit antidepressant effects in animal models [85], and that AMPA receptor activation has been suggested to be involved in the antidepressant actions of ketamine and riluzole [86, 87], which are effective antidepressant treatments, this mechanism is of interest in terms of the efficacy of mGlu2/3 receptor antagonists for the treating depression in the clinic.
3. 3. mGlu5 Receptor Antagonists
The antidepressant potentials of mGlu5 receptor antagonists have been investigated using selective allosteric negative modulators, such as 6-methyl-2-(phenylethynyl)-pyridine (MPEP) and 3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]-pyridine (MTEP). Both compounds reduced immobility times in despair models, such as forced swim and tail suspension tests [88-90]. The antidepressant effects of MPEP in the forced swim test were no longer observed in knockout mice lacking mGlu5 receptors [88], indicating that these compounds exerted their effects via the mGlu5 receptor. Moreover, subchronic administration of MPEP or MTEP attenuated abnormal behaviors (learning deficits in a passive avoidance paradigm or locomotor hyperactivity in a novel environment) induced by olfactory bulbectomy [89, 91]. Molina-Hernandez et al. [92] reported that MTEP injection into the lateral septal nuclei increased reinforced lever presses and caused a comprehensive rightward shift of the inter-response time, as observed with desipramine in the differential-reinforcement-of-low-rate 72-s paradigm, thus providing additional evidence for mGlu5 receptor antagonists for antidepressant treatment.
The neuronal mechanisms underlying the antidepressant effects of mGlu5 receptor antagonists have not been fully elucidated. Interactions between the mGlu5 and NMDA receptors have been proposed to be involved in the actions of mGlu5 receptor antagonists. As described in [2.3], the mGlu5 receptor positively regulated NMDA receptor function. Thus, mGlu5 receptor blockade may have a negative impact on NMDA receptor function. This is interesting, as NMDA receptor antagonists like ketamine have been proven to be a very effective approach for treating depression in both animal models and clinical studies [71, 72, 87]. Moreover, mGlu5 receptor activation on astrocytes increases glutamate release from glial cells in the nucleus accumbens, and glutamate released from glial cells selectively activates extrasynaptic NR2B-containing NMDA receptors, but not intrasynaptic NR2A-containing NMDA receptors [93]. Because increased glutamate release from glial cells, which leads to stimulation of extrasynaptic NMDA receptors, has been considered to be among the causes of depression, indirect extrasynaptic NMDA receptor blockade by mGlu5 receptor antagonists could be a very interesting approach to treating depression.
3. 4. mGlu7 Receptor Ligands
Roles for the mGlu7 receptor in depression have been studied, although its involvement is rather complicated. Knockout mice lacking mGlu7 receptors displayed antidepressant-like phenotypes in a forced swim test and a tail suspension test without significant changes in locomotor activities [94]. mRNA expression levels of the glucocorticoid and 5-HT1A receptors in the hippocampus increased in mGlu7 receptor-null mice [95], suggesting that a feedback mechanism within the HPA axis may be enhanced. In line with this observation, mGlu7 receptor-null mice showed hypersensitivity to dexamethasone, as demonstrated by a marked serum corticosterone reduction in response to dexamethasone challenge [95]. This is opposite to what is seen in many depressed patients who have a blunted response to dexamethasone challenge [96].
In contrast, N,N'-dibenzhydrylethane-1,2-diamine dihydrochloride (AMN082), an allosteric mGlu7 receptor agonist, exhibited antidepressant effects in forced swim and tail suspension tests [97]. The antidepressant effect of AMN082 was not observed in knockout mice lacking mGlu7 receptors, showing that AMN082 exerted its effect through mGlu7 receptor activation. Contradictory findings between stimulation and blockade of this receptor remain to be resolved.
Although not statistically significant, results with mGlu7 receptor-null mice showed increased locomotor activity, which may affect the outcomes in the forced swim test. In addition, the mGlu7 receptor undergoes rapid internalization upon stimulation with AMN082 [98], which, in turn, may lead to mGlu7 receptor blockade. There are several issues to be resolved regarding the neuronal mechanisms by which mGlu7 receptor affects depressive states, including its sites of action and relationships with other glutamate receptors.
4. CHEMISTRY
4.1. mGlu2/3 Receptor Agonist/mGluR2 Receptor Potentiators
In initial studies investigating the functions of mGlu2/3 receptors, L-CCG-I, ACPD and DCG-IV were used as non-selective ligands and conformationally restricted glutamate analogs (Fig. 1) [99-101]. LY354740 was described in 1997 as the first potent and selective agonist for mGlu2/3. It activates the mGlu2 (EC50 = 5 nM) and mGlu3 (EC50 = 24 nM) receptors in vitro and has potent pharmacological effects in animals [102]. LY354740 is a comformationally restricted bicycloamino acid, (1S,2S,5R,6S)-2-aminobicyclo[3.1.0]hexane-2,6-dicarboxylic acid. For this type of bicyloamino acid, in the 2-position of the bicyclohexane ring, LY379268 has an oxygen atom, LY389795 has a sulfur atom, and MGS0028 has an oxo group [23, 103]. These compounds have been reported to have highly potent mGlu2/3 receptors agonist activities.
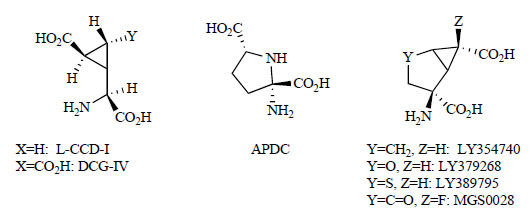
Competitive mGlu2/3 receptor agonists.
Two compound series have been reported as mGlu2 receptor allosteric potentiators (Fig. 2). The first series includes an allosteric modulator of the mGlu2 receptor from an N-aryl pyrinin-3-ylmethylsulfonamide derivative, 4-MPPTS [104]. In this compound type, cyPPTS is a very potent potentiator (24 nM) [105]. Another series of compounds results from 4’-alkoxyacetophenone derivatives, and is typified by compound 1. Compound 1 is a selective mGlu2 receptor potentiator (300 nM) with no allosteric activities against mGlu1, 3, 4, 5, 7 or 8 [106, 107].
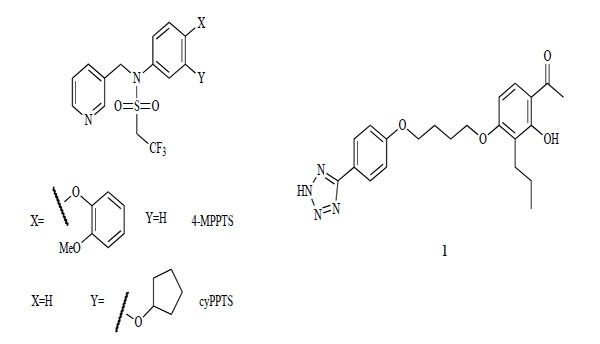
mGlu2 receptor potentiators.
4.2. mGlu2/3 Receptor Antagonists
Compared with other receptor subtypes, mGlu2/3 receptor antagonists have rarely been reported in the literature. In 1996, a glutamate analog, LY341495, was disclosed as a selective competitive mGlu 2/3 receptor antagonist in a patent application (Fig. 3) [108]. LY341495 has nanomolar affinities for mGlu 2/3 receptors [109, 110]. MGS0039 containing a bicyclo[3.1.0]hexane ring system was reported to have nanomolar affinities for mGlu 2/3 receptors [111]. The series of mGlu 2/3 receptors antagonists that have amino acid moieties are found at low levels in the CNS. In fact, ester prodrugs of MGS0039 are reported to deliver MGS0039 after hydrolytic release of the ester prodrugs [112].

Antagonists of mGlu 2 receptor.
New classes of mGlu 2 antagonists without amino acid moieties have been identified. Triazole derivative 2 was reported as an antagonist with moderate affinity for the mGlu 2 receptor (Ki = 0.1 μM) [113]. A series of patents were published on benzodiazepine derivative 3, which reportedly had a very low nanomolar affinity for the mGlu 2 receptor [114]. Thiazolopyrimidine derivative 4 and pyrrazolopyrimidine derivative 5 represented novel structural templates for mGlu 2 receptor antagonists [115 – 119]. Thiazolopyrimidine derivatives showed moderate binding affinity for the mGlu 2 receptor, while pyrrazolopyrimidine had low nanomolar affinity. Recently, a structurally different mGlu 2 receptor antagonist, imidazole 6, was disclosed (binding affinity for mGlu 2 receptor (IC50= 7nM) [120]. These new classes of mGlu 2 receptor antagonists have limited disclosures regarding the details of their subtype selectivities, their modes of binding, including activities as competitive and non-competitive modulators, binding sites, and their pharmacology.
4.3. mGlu1 Receptor Antagonists
Competitive mGlu1 receptor antagonists are poorly represented in the literature. Fig. (4) shows some selective competitive mGlu1 receptor antagonists. These antagonists are analogs of glutamine and phenylglycine. (S)-4-CPG, a phenylglycine analog, is a selective antagonist with weak effects for the mGlu2 receptor [121]. The 2-Methyl analog of (S)-4-CPG, LY367385, is more potent and more selective than (S)-4-CPG for the mGlu1 receptor, without any effects for the group II mGlu receptors [122]. AIDA, a conformationally restricted analog between the phenyl group and an amino acid carbon, selectively antagonizes the mGlu1 receptor, with weak effects for the mGlu5 and 2 receptors [123, 124]. Compound 7, with a replacement in the phenyl ring of (S)-4-CPG with cuban is a potent, selective antagonist for the mGlu1 receptor [125, 126]. Since competitive mGlu1 receptor antagonists show poor potencies, selectivities, and levels of CNS exposure, they have not yet become clinically useful therapeutic agents [127, 128].
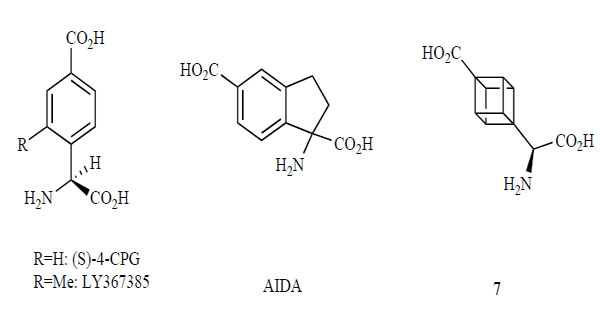
Competitive mGlu1 receptor antagonists.
Negative allosteric mGlu1 receptor modulators are structurally different from glutamate and phenylglycine. Several different structures are shown in Fig. (5.) CPCCOEt was first identified as a negative allosteric modulator in the late 1990s [129-131]. Then, several new classes of mGlu1 receptor antagonists that were structurally distinct from CPCCOEt were identified. BAY36-7620, containing butyrolactone, is a potent (IC50 = 160 nM) and selective antagonist for the mGlu1 receptor. It inhibits >60% of mGlu1a receptor constitutive activity (IC50 = 380 nM). Thus, BAY36-7620 was the first mGlu1 receptor inverse agonist to be described [132]. At about the same time, some different types of heteroaromatic compounds were reported. Compound 8 containing an indole ring shows a low micromole affinity for the mGlu1 receptor, and pyridine ring fused indole derivatives 9 is also a low affinity mGlu1 receptor antagonist [133, 134]. A series of patents were published on YM-29818 containing a thiazolobenzimidazole ring [135-137]. The specificity of YM-29818 for the mGlu1 receptor (IC50 = 16 nM) included subtypes 2 to 7, as well as several other receptors, transporters, and ion channel targets. From in vivo experiments, orally administered YM-298198 showed a significant analgesic effect in streptozotocin-induced hyperalgesic mice (30 mg/kg) [138]. In 2006, in a series of tricycloaromatics, novel pyridothienopyrimidine derivatives were reported as mGlu1 receptor antagonists, such as 10, that had IC50 values of <50 nM [139].
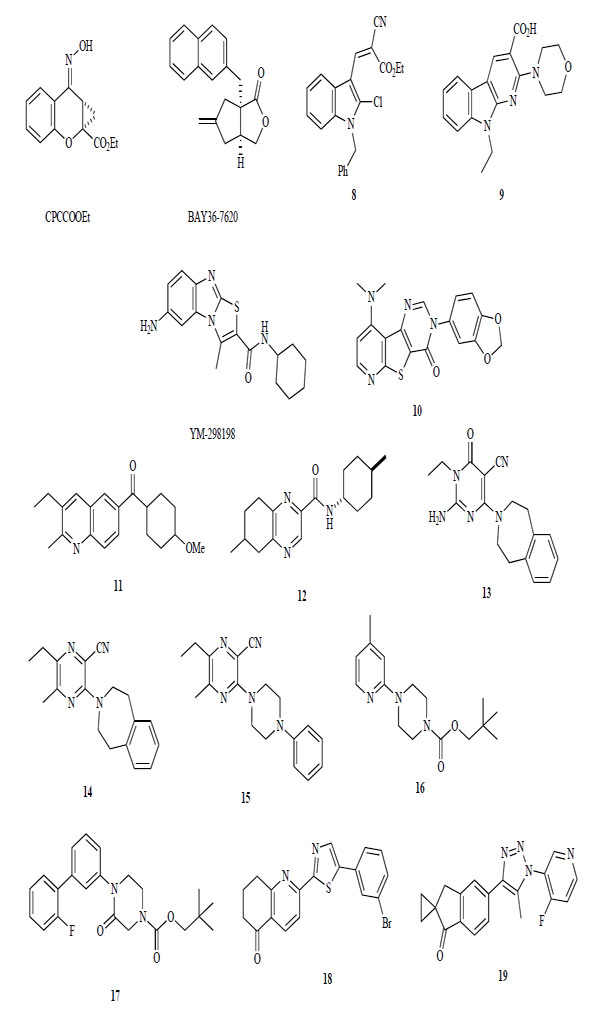
Non-competitive mGlu1 receptor antagonists.
Quinoline and quinoxaline derivatives, such as 11 and 12, were found to be mGlu1 receptor antagonists. Compound 11 exhibited high activity for the mGlu1 receptor (IC50 = 2.97 nM) [140]. Compound 12 had good affinity for the mGlu1 receptor (IC50 = 52n M) and no affinity for mGlu5 receptor (IC50 = 7289 nM) [141].
A series of heteroaromatics and cycloamine derivatives, 13–19, were also published. Benzoazepine and pyrimidine derivative 13 has an IC50 value of 3 nM for binding affinity to the mGlu1 receptor, an IC50 value of 9 nM [142]. Pyrazine analog 14 also shows high binding affinity for the mGlu1 receptor (IC50 = 6 nM) [143]. Piperazine derivative 17, modified from benzoazepine moiety in 14 to phenyl-piperazine, is a potent negative allosteric modulator of the mGlu1 receptor (IC50 = 23 nM) [144]. In 2006, N-pyridyl- and N-phenyl-piperazine derivatives were identified. Compounds 16 and 17 had reported affinities for the mGlu1 receptor of 6.9 nM and 38 nM, respectively, and antagonized the increase of locomotor activity in rats with amphetamine at a dose of 30 mg/kg [145, 146].
Recently, novel diaryl substituted 5-membered heteroaromatic derivatives 18 and 19 were disclosed. Compound 18 exhibited an IC50 value of 2.3 nM and activity against the disruption of prepulse inhibition in methamphetamine treated rats at 1–10 mg/kg [147, 148].
4.4. mGlu5 Receptor Agonists/mGlu5 Receptor Potentiators
3,5-DHPG was identified as the first group I receptor selective agonist (Fig. 6). However, 3,5-DHPG lacks subtype selectivity and has low potency [149, 150]. In 1999, compounds 20 and 21 were described as selective mGlur5 receptor agonists. Their EC50 values were 11 nM and 49 nM, respectively [151]. Their apparent higher potency and selectivity for mGlu5 receptor might make these compounds important new pharmacological tools. Despite these significant advances, highly potent and subtype-selective competitive mGlu receptor agonists have not been identified.
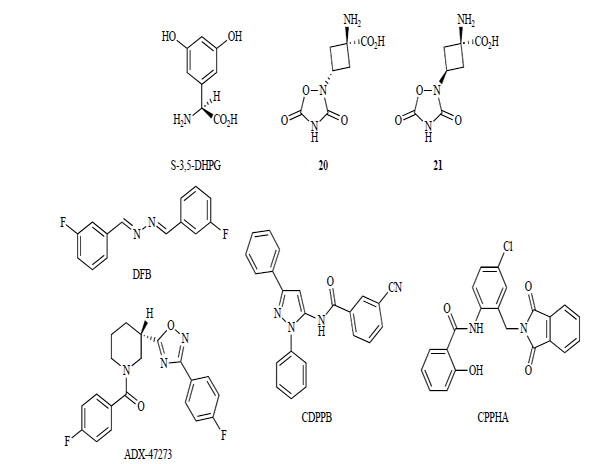
mGlu5 receptor agonists.
In an attempt to overcome the low potency, low selectivity, and poor blood-brain barrier permeation of most mGlu receptor ligands, investigations of non-competitive mGlu5 receptor modulators began in the mid-1990s. mGlu5 receptor positive allosteric modulators began to be developed after 2000. Three different structural types of mGlu5 receptor positive allosteric modulators were reported: DFB, CDPPB, and CPPHA [152-154]. These modu-lators do not affect ligand binding to the orthosteric glutamate binding site, but potentiate its response to glutamate. DFB and CDPPB bind to the MPEP site and displace binding of radioligand to a well-characterized binding site for MPEP [155, 156]. Thus, some reports indicate that these compounds have antipsychotic-like effects [156, 157].
In 2005, a fourth structural series, ADX-47273, was reported by Addex pharm with similar properties [158, 159].
4.5. mGlu5 Receptor Antagonists
The first mGlu 5 receptor antagonists were identified from agonists having either a glutamate or a phenylglycine moiety (Fig. 7). These antagonists bind competitively at the glutamate-binding site. These early competitive antagonists lacked subtype selectivity. For example, phenyglycine derivative 22 showed weak antagonist activity for human mGlu5 receptor, as well as antagonist activity for the mGlu1 receptor [121].
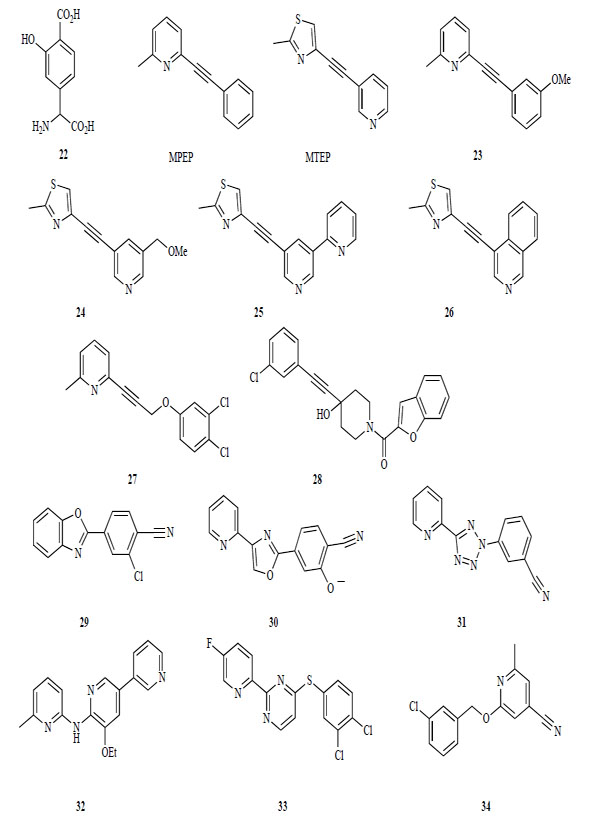
Antagonists of mGlu 5 receptor.
In contrast, the most widely known pharmacological tool for showing the therapeutic potential of mGlu5 receptor antagonists is MPEP. Both MPEP and the structurally related MTEP are highly selective antagonists for the mGlu1 receptor [158-160]. These compounds are structurally distinct from glutamate and bind to an allosteric site in the transmembrane domain. After identifying MPEP and MTEP, many derivatives were disclosed in patents and in the literature. Several substitutions were introduced into the 3-position of the phenyl ring of MPEP and the 5-position of the pyridine ring of MTEP.
Compound 23, a derivative with a methoxy group in the 3-position of the benzene ring of MPEP, shows a similar potency for the mGlu5 receptor as MTEP. The methoxy group of 23 can be easily radiolabeled by a reaction of the phenolic intermediate with [3H]-methyl iodide. This radioligand with high specific activity and radiochemical purity is useful for labeling the mGlu5 receptor [161]. Introduction of a methoxymethyl group into the 5-pyridyl position of MTEP gives 24 with a similar potency to MTEP [162]. A radioactive compound of 24 has also been synthesized and used for in vitro binding assays using either rat brain tissues or cells that express recombinant human mGlu5 receptors [163].
Additional modifications of the pyridine ring of MTEP were investigated. Introduction of pyridine at the 5-pyridyl position of MTEP and transformation of the isoquinoline ring from the pyridine ring of MTEP gave 25 and 26, respectively. These compounds show high in vitro potencies (25, IC50 = 22 nM, Ki = 28 nM; 26, IC50 = 20 nM) [164, 165]. Further explorations of mGlu5 receptor agonists by high-through-put-screening and optimization led to a novel series of pyridinyl-alkyne derivatives that were structurally related to MPEP (25: IC50 = 15 nM) [166]. Recently, a novel phenyl-alkyne derivative was disclosed as an mGlu5 receptor antagonist (28: IC50 = 290 nM) [167].
Aryl benzoxazole 29 was identified by high-through-put-screening as a potent mGlu5 receptor antagonist [168]. Compound 30 containing an oxazole ring substituted with an aryl group was disclosed in a patent [169]. However, 29 was not suitable for in vivo studies due to its poor solubility and poor pharmacokinetic profile (oral bioavailability F = 0.7 %) [168]. Compound 29 was modified based on SAR studies of the common structural features of 29, oxazole derivative 30, MPEP and MTEP to reveal a series of azole derivatives containing a pyridine and 1 or 2 phenyl rings, such as 31 [170].
Recently, some new classes of mGlu 5 receptor antagonists, dipyridyl amine 32, thiopyrimide 33, and arylmethoxypyrimide derivative 34, were identified by high-through-put screening (32; IC50 = 12 nM, 33; IC50 = 80 Nm 34; Ki = 1 nM). Compounds 32 and 34 have low to moderate oral bioavailabilities (32; F = 38 %, 34; F = 4.7 %).
4.6. mGlu7 Receptor Ligands
To date, mGlu7 ligands are poorly represented in the literature. Only a mGlu7 receptor-selective agonist has been described, AMN082 (Fig. 8) [171, 172]. This compound was identified using high-throughput screening as the first full allosteric agonist for the mGlu7 receptor. This compound is structurally unrelated to any known mGlu receptor ligand. AMN082 has a unique mechanism of action by fully activating mGlu7 receptor through an allosteric site far removed from the glutamate-binding pocket [98].

mGlu7 receptor ligands.
There are only a limited number of selective mGlu7 receptor antagonists. MDIP was identified by high- throughput screening, and MMPIP was synthesized by chemical modification of the phenyl group in the isoxazole ring system to a pyridyl group. In a cAMP assay, MDIP and MMPIP had IC50 values of 99 nM and 220 nM, respectively. The inhibitory modes of these compounds are non-competitive and allosteric, and MMPIP appears to have an inverse agonist activity [173].
CONCLUSIONS AND FUTURE DIRECTIONS
Accumulating evidence indicates that mGlu receptors are significantly involved in psychiatric disorders and may serve as intriguing targets for drug discovery for the treatment of psychiatric disorders (Table 2). Among the mGlu receptors, mGlu2/3 receptor agonists have been proven to be effective for the treatment of not only schizophrenia, but also certain anxiety disorders in clinical settings. LY354740 was shown to exert anxiolytic effects in a fear-potentiated startle model in human volunteers [7], and LY354740 or LY544344, a prodrug of LY354740, showed efficacy in human anxiety models (panic provocation induced by the intravenous injection of cholecystokinin tetrapeptide or CO2 challenge) [8, 9]. Therefore, together with the recent remarkable find-ings that LY2140023, a prodrug of the mGlu2/3 receptor agonist LY404039, provided improvements for both positive and negative symptoms of schizophrenia, mGlu2/3 receptor stimulation has paved the way for developing novel approaches beyond monoamine therapy for the treatment of psychiatric disorders, such as schizophrenia and anxiety.
Possible Therapeutic Indications for Agents Acting on mGlu Receptors
| Group | Subtype | Possible Therapeutic Indications | |
|---|---|---|---|
| Group I | mGlu1 | agonists/PAMs | cognitive disorders |
| antagonists | anxiety, schizophrenia, pain | ||
| mGlu5 | agonists/PAMs | schizophrenia, cognitive disorders | |
| antagonists | depression/anxiety, pain, drug addiction, gastroesphageal reflux disease, Parkinson's disease, migrain | ||
| Group II | mGlu2 | agonists/PAMs | schizophrenia, anxiety |
| antagonists | depression/anxiety, cognitive disorders | ||
| mGlu3 | agonists/PAMs | schizophrenia | |
| antagonists | ? | ||
| Group III | mGlu4 | agonists/PAMs | Parkinson's disease, anxiety (?) |
| antagonists | ? | ||
| mGlu7 | agonists/PAMs | depression/anxiety (?) | |
| antagonists | depression/anxiety (?) | ||
| mGlu6 | agonists/PAMs | ? | |
| antagonists | ? | ||
| mGlu8 | agonists/PAMs | depression/anxiety (?) | |
| antagonists | ? |
Preclinical evidence is accumulating on the efficacy of mGlu2/3 receptor antagonists (depression), mGlu5 receptor agonists/potentiators (schizophrenia) and mGlu5 receptor antagonists (depression) in several animal models. Although a large number of patents for these targets have been published, there are few compounds that have entered clinical trials. Human proof-of-concept studies with compounds acting on the above mGlu receptors are necessary in order to address the utility of these mGlu receptors for treating psychiatric disorders.

