All published articles of this journal are available on ScienceDirect.

α7 Nicotinic Receptor Agonists: Potential Therapeutic Drugs for Treatment of Cognitive Impairments in Schizophrenia and Alzheimer’s Disease
Abstract
Accumulating evidence suggests that α7 nicotinic receptors (α7 nAChRs), a subtype of nAChRs, play a role in the pathophysiology of neuropsychiatric diseases, including schizophrenia and Alzheimer’s disease (AD). A number of psychopharmacological and genetic studies shown that α7 nAChRs play an important role in the deficits of P50 auditory evoked potential in patients with schizophrenia, and that (α nAChR agonists would be potential therapeutic drugs for cognitive impairments associated with P50 deficits in schizophrenia. Furthermore, some studies have demonstrated that α7 nAChRs might play a key role in the amyloid-β (Aβ)-mediated pathology of AD, and that α7 nAChR agonists would be potential therapeutic drugs for Aβ deposition in the brains of patients with AD. Interestingly, the altered expression of α7 nAChRs in the postmortem brain tissues from patients with schizophrenia and AD has been reported. Based on all these findings, selective α7 nAChR agonists can be considered potential therapeutic drugs for cognitive impairments in both schizophrenia and AD. In this article, we review the recent research into the role of α7 nAChRs in the pathophysiology of these diseases and into the potential use of novel α7 nAChR agonists as therapeutic drugs.
α7 NICOTINIC ACETYLCHOLINE RECEPTORS
(S)-(-)-Nicotine (Fig. 1, hereafter simply nicotine), the main addictive component of tobacco, activates and desensitizes nicotinic acetylcholine receptors (nAChRs). In the central nervous system (CNS), nAChRs normally respond to acetylcholine (ACh) (Fig. 1) and modulate neuronal excitability and synaptic communication [1-4]. The health consequences of smoking and the mechanisms involved in nicotine dependence continue to be subjects of great interest [5-9]. In addition, recent attention has been directed to the potential role of nAChRs in diseases and therapeutic targets [10-23].
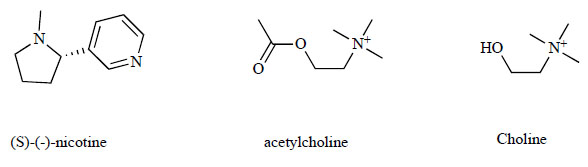
Chemical structures of (S)-(-)-nicotine, acetylcholine (ACh) and choline.
The nAChRs are ligand-gated ion channels that are distributed throughout the human CNS and that each consists of five subunits (a combination of α and β subunits). At present, nine α (α2-α10) and three β (β2-β4) subunits have been identified and cloned in humans. The numerous combinations of α and β subunits or of α subunits alone can generate many subtypes of nAChRs with different physiologies, pharmacologies and anatomical distributions [4]. Two major subtypes exist in the brain, those comprised of α4β2 and those comprised of α7 subunits. The former contribute >90% of the high affinity binding sites for nicotine in the rat brain [24]. The low affinity binding sites (α7 subunits) for nicotine are recognized by their nanomolar affinity for α-bungarotoxin [25]. The α7 nAChRs have an unusually high permeability to Ca2+ compared to other subtypes and exhibit exceptionally rapid desensitization following exposure to agonists [8, 26-30]. Choline (Fig. 1) is an essential physiological component of the cerebrospinal fluid (CSF) and is important for the structural integrity of cell membranes, ACh synthesis, and lipid and cholesterol transport and metabolism. Several lines of evidence suggest that choline is a full agonist of α7 nAChRs, but not other nAChR subtypes [31-34].
The α7 nAChRs are assumed to comprise five α7 subunits and to differ from other subtypes of nAChRs. In particular, the α7 nAChRs may play a distinct role in regulating neuronal plasticity. By elevating intracellular Ca2+ levels in discrete neuronal locations, these ligand-gated ion channels may influence numerous physiological processes in the developing and adult CNS [4, 23, 26, 27, 35]. Several lines of evidence suggest that both pre- and postsynaptic α7 nAChRs modulate neurotransmitter release in the brain through Ca2+-dependent mechanisms, and that the α7 nAChRs play a role in regulating neuronal growth and differentiation in the developing CNS [4, 26, 35-38]. Furthermore, it has been proposed that intracellular Ca2+ may be coregulated by N-methyl-D-aspartate (NMDA) receptors and α7 nAChRs in the brain [26]. Together with NMDA receptors, postsynaptic α7 nAChRs may serve to regulate intracellular Ca2+ levels in neurons, whereas presynaptic α7 nAChRs could serve as a feedback mechanism for modulating glutamatergic transmission. Thus, it is possible that a close interaction between cholinergic and glutamatergic pathways, mediated by α7 nAChRs and NMDA receptors, may play a role in the pathophysiology of neuropsychiatric diseases such as schizophrenia and Alzheimer’s disease (AD) [4, 23, 26, 35-41].
In this article, we review the role of α7 nAChRs in the pathophysiology of neuropsychiatric diseases such as schizophrenia and AD. In addition, we also review recent advances in the potential use of α7 nAChR agonists as therapeutic drugs for these diseases.
SCHIZOPHRENIA AND α7 nAChRs
The binding sites (α7 subtype) labeled by [125I]α-bungarotoxin are different from the high affinity binding sites (α4β2 subtype) for nicotine [42]. In the hippocampus, [125I]α-bungarotoxin binds most intensely to inhibitory interneurons in the CA3 region of Ammon’s horn and in the hilus of the dentate gyrus [43]. The authors of the latter study subsequently suggested that inhibitory interneurons with α7 nAChRs are possible candidates for medication of the habituation of auditory responses in the hippocampus because activation of the interneurons via α7 nAChRs would increase the inhibitory synaptic input to pyramidal neurons and thereby diminish the responsiveness of these pyramidal neurons to sensory stimulation [44]. This parallels studies of postmortem human tissue that documented a decreased expression of hippocampal α7 nAChRs in schizophrenic patients [45]. It has been shown that [125I]α-bungarotoxin binding is reduced in the thalamic reticular nucleus of schizophrenic subjects [46], and that α7 nAChR protein levels are reduced in the frontal cortex in patients with schizophrenia [47]. Furthermore, Marutle et al. [48] observed a reduction of α7 nAChRs but an increase of [3H]cytisine binding to α4β2 nAChRs in the cingulate cortex of schizophrenic patients. Thus, it seems that schizophrenic patients have fewer α7 nAChRs in the hippocampus, a condition which may lead to failure of cholinergic activation of inhibitory interneurons, clinically manifested as decreased gating of the response to sensory stimulation [23, 37, 44, 49, 50].
In regard to the search for peripheral biological markers for schizophrenia, Perl et al. [51] investigated the mRNA levels of α7 nAChRs in peripheral blood lymphocytes of schizophrenic patients and healthy controls. They found a significant reduction of α7 nAChR mRNA levels on lymphocytes of schizophrenic patients [51]. This reduction was not a result of medication, because the non-medicated patients displayed the same levels of reduction in α7 nAChR mRNA. In addition, the possibility that the observed decrease in α7 nAChR mRNA levels resulted from nicotine consumption in smoking was excluded, because healthy smokers exhibited the same levels of α7 nAChR mRNA as non-smokers [51]. These findings suggest that mRNA levels of α7 nAChRs in peripheral blood lymphocytes may serve as a reliable peripheral biological marker for schizophrenia.
SENSORY GATING DEFICITS IN SCHIZOPHRENIA AND α7 nAChRs
Our knowledge of the deficits of sensory gating in schizophrenia derives from the clinical observation that patients report failures of information processing characterized by poor sensory gating [44, 52-54]. The underlying problem is evident in the inability of people with schizophrenia to adequately filter their response to incoming sensory stimulation, as measured by their inhibitory processing of the P50 auditory evoked potential. The P50 auditory evoked potential is a positive electroencephalographic waveform that occurs 50 msec after presentation of an auditory stimulus. When pairs of auditory stimuli are presented, with a 500 msec interstimulus interval, schizophrenic patients fail to adequately inhibit the P50 response to the second stimulus. Normal subjects, however, have significantly reduced responses to the second stimulus [44, 52, 54, 55]. The reduced response to the second stimulus reflects inhibitory processing of the information that may function to protect the individual from being overwhelmed by incoming, repetitive sensory information. It is known that nicotine transiently normalizes the deficits in P50 auditory evoked potential in schizophrenic patients [56, 57].
Genetic linkage analysis of deficits in P50 auditory evoked potential in families of patients with schizophrenia has revealed a peak LOD score at 15q13-q14, and the LOD score was 5.3 (theta=0.00) at the D15S1360 marker, which is located in intron 2 of the gene for the α7 nAChR subunit (CHRNA7) [58]. The CHRNA7 gene is located on chromosome 15q13-q14, a region linked with schizophrenia in several earlier studies [59, 60]. The CHRNA7 has a partial duplication of exons 5-10, including the intervening introns (CHRFAM7) that map approximately 0.5 Mb proximal to the full-length CHRNA7 gene [61]. The D15S1360 microsatellite repeats, in intron 2 of the CHRNA7 gene, cosegregate with an auditory gating deficit in family linkage studies of schizophrenic patients [58]. Furthermore, in a mutation screening of the CHRNA7 gene from schizophrenics and controls, Leonard et al. [62] identified the promoter polymorphisms that decreased the subunit transcription and P50 inhibition in schizophrenia. Moreover, an association has been demonstrated between the homozygous 113 bp allele on the D15S1360 polymorphism of the CNRNA7 gene and smoking risk in schizophrenia [63]. Taken together, these results suggest that the CHRNA7 gene is likely susceptible to the deficits of P50 sensory gating in schizophrenia [10, 23, 44, 49, 50, 53, 58, 63-67]. Interestingly, a 2 bp deletion in exon 6 of CHRFAM7A, which disrupts the hybrid gene for the CHRNA7 gene, was associated with episodic memory performance in schizophrenia, suggesting that the CHRFAM7A/CHRNA7 locus plays a role in modulating episodic memory function [68].
Taken together, these results suggest that α7 nAChRs likely play an important role in the deficits of P50 auditory evoked potential in patients with schizophrenia, and that α7 nAChR agonists would be potential therapeutic drugs for cognitive impairments associated with P50 deficits in schizophrenia [10, 23, 64, 65, 69, 70].
ALZHEIMER’S DISEASE (AD) AND α7 nAChRs
Identification of the loss of cholinergic neurons in the basal forebrain and of cholinergic innervations of the cerebral cortex in Alzheimer’s disease (AD) [71-73] was followed by investigations into the involvement of cholinergic receptors. With regard to α7 nAChR protein expression, the investigations show conflicting results. For example, it has been reported that there is no significant relation between [125I]α-bungarotoxin binding in the frontal cortex and AD [74, 75]. Although an early study of Davies and Feisullin [74] reported a 40% reduction in [125I]α-bungarotoxin binding in the temporal cortex of patients with AD, this was not replicated [76]. The results of most studies show no significant [77, 78] or minor [79] loss of α7 subunit protein levels in the temporal cortex of patients with AD, although a reduction has been demonstrated in the frontal cortex of such patients [80]. In the hippocampus, a 25% reduction in [125I]α-bungarotoxin binding (as revealed using membrane homogenate) was observed in patients with AD relative to age-matched control subjects, although the number of control cases studied was small [76]. However, an autoradiographic investigation did not detect significant changes in [125I]α-bungarotoxin binding in a large series of patients with AD, but rather, in common with Burghaus et al. [79], a considerable variation in the density of the α7 receptor subtype was seen among patients with AD [81]. Such variation in α7 nAChR expression may reflect disease severity, and it should be evaluated in cohorts of patients with detailed clinical and pathological assessments. Despite a reported 36% reduction in α7 protein expression in the hippocampal formation of patients with AD [77], a 65% increase in α7 subunit mRNA expression has also been demonstrated in patients with AD [76], suggesting the possibility of a compensatory mechanism. Recently, Counts et al. [82] reported that α7 nAChR was up-regulated in cholinergic basal forebrain neurons in patients with AD. In the same study, dysfunction of the basocortical cholinergic projection neurons of the nucleus basalis (NB) was correlated with cognitive deficits in AD. The authors found that cholinergic NB neurons displayed a statistically significant up-regulation of α7 nAChR mRNA expression in subjects with mild to moderate AD compared with those with no cognitive impairment (NCI) or mild cognitive impairment (MCI) (P<0.001). The expression levels of α7 nAChRs were inversely associated with the Global Cognitive Score and with Mini-Mental State Examination performance. Very recently, Ikonomovic et al. [83] found that increased α7 nAChR binding of [3H]methyllycaconitine (MLA)( Fig. 2) was associated with diagnosis of AD based on the National Institute on Aging-Reagan Institute criteria (P=0.02) and, albeit weakly, the presence of cortical β-amyloid (Aβ) plaque in AD (P=0.08). Taken together, these results suggest that the up-regulation of α7 nAChR may occur as a compensatory mechanism to maintain neuronal function during AD progression [83]. Furthermore, it has been reported that genetic variation (rs6494223) in the CHRNA7 gene is associated with delusional symptoms in AD [84], suggesting that α7 nAChRs may be a suitable target for the treatment of AD with psychosis. In addition, patients with Alzheimer's disease have also been reported to show deficits of P50 auditory sensory gating relative to controls [85]. It has also been suggested that the disturbed sensory gating in patients with AD might result from cholinergic dysfunction or α7 nAChR loss.
Chemical structure of MLA.
β-AMYLOID AND α7 nAChRs
β-Amyloid (Aβ) binds with high affinity to neuronal α7 nAChRs [86, 87]. This interaction leads to intraneuronal accumulation of Aβ1-42 (Aβ42)– α7 nAChR complexes [88], rapid tau phosphorylation [89], severe impairment of α7 nAChR channels [90, 91], cholinergic neurotransmission defects [92], and neuronal cell death [86]. These findings indicate that chronic perturbation of the α7 nAChRs by Aβ42 in AD brains could cause neuronal dysfunction and neurodegeneration, resulting in the accumulation of Aβ-rich amyloid plaques and phosphorylated tau-containing neurofibrillary tangles (NFTs). Recently, Dziewczapolski et al. [93] showed that, despite the presence of high amounts of amyloid precursor protein (APP) and amyloid deposits, deleting the α7 nAChR subunit in the mouse model of AD leads to protection from dysfunction in synaptic integrity (pathology and plasticity) and learning and memory behavior. Hence, disrupting the Aβ42–α7 nAChR interaction may represent a novel approach to reducing Aβ42-mediated functional deficits, neurodegeneration, and possibly the clinicopathological features of AD.
Taken together, these results suggest that α7 nAChRs are likely to play a key role in the Aβ-mediated pathology of AD, and that α7 nAChR agonists would be potential therapeutic drugs for AD [39, 40, 93, 94].
α7 NICOTINIC RECEPTOR AGONISTS
3-(2,4)-Dimethoxybenzilidine Anabaseine (DMXB-A; Also Known as GTS-21)
While there are a number of nicotinic receptor agonists known to be selective for the α4β2 subtype, there are some agonists which bind the α7 nAChRs selectively over other subtypes [13, 18, 20]. Anabaseine (2-(3-pyridyl)-3,4,5,6-tetrahydropyridine) (Fig. 3), a naturally occurring substance in nemertines, is an agonist at the neuromuscular junction [95] and is structurally related to nicotine. The better known compound anabasine (neonicotine; 3-(2-piperidinyl)pyridine) (Fig. 3) is a weak nicotinic alkaloid found in tobacco that lacks the imine double bond present in anabaseine. Three analogues of anabaseine, 3-(2,4)-dimethoxybenzilidine anabaseine (DMXB-A; also known as GTS-21), 3-(4)-dimethylaminobenzylidine anabaseine (DMAB), and 3-(4)-dimethylaminocinnamylidine (DMAC), have been reported to be functionally selective for the α7 nAChRs [96] (Fig. 3). Compared with anabaseine and the other derivatives, DMAC was found to be the most potent at displacing [125I](-bungarotoxin binding (putative α7 subtype) and the least potent at displacing [3H]cytisine binding (putative α4β2 subtype) to brain membranes. These anabaseine derivatives were partial agonists at α7 nAChRs [96]. Furthermore, DMXB-A bound to human α4β2 nAChRs (Ki=20 nM) 100-fold more potently than to human α7 nAChRs, and was 18- and 2-fold less potent than (–)-nicotine at human α4β2 and α7 nAChRs, respectively [97]. The primary human metabolite, 3-(4-hydroxy-2-methoxybenzylidine) anabaseine (4-OH DMXB-A; Fig. 3), of DMXB-A exhibited a similar level of efficacy for human α7 nAChRs [98, 99].
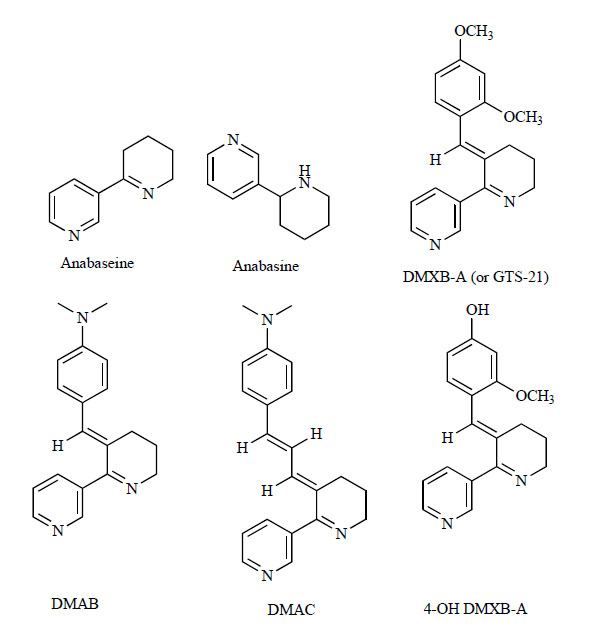
Chemical structures of anabaseine, anabasine, DMXB-A, DMAB, DMAC and 4-OH DMXB-A.
Initial (phase I) clinical studies on DMXB-A have been reported [100]. DMXB-A was administered to 87 healthy volunteers. Initially, the effects of single doses (range, 1-250 mg) were assessed. The elimination half-life ranged between 0.5 and 1.0 h for DMXB-A and its major phase I metabolite, 4-OH DMXB-A. No serious adverse effects were reported at these doses. At twice daily doses of 75 and 150 mg for 5 days, DMXB-A improved the cognitive function of young adult volunteers. Furthermore, DMXB-A improved long-term memory as well as working memory and attention, as measured by the Cognitive Drug Research test battery [100]. A randomized, placebo-controlled, multiple dose study of the safety, pharmacokinetics and cognitive effects of DMXB-A in healthy volunteers was also reported [101]. A total of 18 subjects were randomized to receive DMXB-A (25, 75, and 150 mg) or a placebo administered three times daily for 5 days with a 10 day washout period between drug-taking periods. DMXB-A was well tolerated up to doses of 450 mg/day, with no clinically significant safety findings. Peak plasma levels (Cmax) were achieved at 1-1.4 h after the first dose and 1-1.2 h after 5 days of dosing. Cmax and the area under the plasma concentration-time curve (AUC) of DMXB-A and metabolite 4-OH-DMXB-A increased in a dose-related manner. DMXB-A showed statistically significant enhancement of three measures of cognitive function (attention, working memory, and episodic second memory) compared to placebo. A relationship between exposure to DMBX-A and the magnitude of the cognitive response was apparent, with a maximal effect observed for doses between 75 and 150 mg three times a day.
Because DMXB-A appeared safe and promising for enhancing cognition, this drug was studied in schizophrenic patients to determined whether the α7 nAChRs activation is responsible for the normalization of the P50 auditory evoked potential deficits in schizophrenia [102]. Additionally, the safety and effects of DMXB-A on neurocognition in schizophrenia patients were also evaluated. DMXB-A was administered in a double-blind, placebo-controlled cross-over design to 12 male and female non-smokers with schizophrenia. DMXB-A was administered orally (150 or 75 mg) followed 2 h later by a half dose (75 or 37.5 mg). DMXB-A improved performance on both the repeatable battery for assessment for neuropsychological status (RBANS) total scale score and the attention scale. DMXB-A also normalized the P50 ratio as well as the test wave amplitude, a more specific measure of inhibition.
On the basis of this initial positive trial, a phase 2 trial was conducted to assess whether the cognitive effects would continue during longer-term administration and whether the clinical ratings would also change [103]. Thirty-one subjects with schizophrenia received DMXB-A at one of two different doses or a placebo for 4 weeks in a three-arm, two-site, double-blind, crossover phase 2 trial. The doses were those used in the phase 1 trial. The Measurement and Treatment Research to Improve Cognition in Schizophrenia (MATRICS) Consensus Cognitive Battery developed by the National Institute of Mental Health was used to assess cognitive effects, and the Scale for the Assessment of Negative Symptoms (SANS) and Brief Psychiatry Rating Scale (BPRS) were used to assess the clinical effects. Subjects continued their current antipsychotic drug during the trial and were nonsmokers. There were no significant differences in the MATRICS cognitive measures between DMXB-A and placebo over the three treatment arms, but patients taking the higher DMXB-A dose experienced significant improvement in the SANS total score and nearly significant improvement in the BPRS total score. Improvement was most notable on the SANS anhedonia and alogia subscales. Examination of the first treatment arm showed effects of DMXB-A on the attention/vigilance and working memory MATRICS domains, compared to baseline. There were, however, increased reports of nausea and restlessness during DMXB-A treatment. Nausea occurred in patients (45%) at the higher dose of DMXB-A, suggesting the known effects of nicotinic agonists on gastrointestinal motility [103]. Considering the high incidence of nausea in the treatment with α7 nAchR agonists, α7 nAchR agonists with 5-hydroxytryptamine-3 (5-HT3) receptor antagonism would be potential therapeutic drugs for cognitive impairments as well as negative symptoms in schizophrenic patients, since the 5-HT3 receptor antagonists have been used for the treatment of nausea [104].
AstraZeneca
Researchers at AstraZeneca reported the profile of AR-R17779, (−)-spiro[1-zabicyclo-[2.2.2]octane-3,5'-oxazolidin -2'-one] (Fig. 4), a potent full agonist of the α7 nAChRs that is highly selective for the α7 subtype over the α74β72 subtypes [105]. AR-R17779 has been widely used as a selective full agonist of the α7 nAChRs. For example, AR-R17779 failed to stimulate locomotor activity in both nicotine-nontolerant and -sensitized rats, whereas nicotine and the putative agonist SIB1765F, [±]-5-ethynyl-3-(1-methyl-2-pyrrolidiny-l)pyridine fumarate (Fig. 4) [100], of α4β2 nAChRs increased the activity under both experimental conditions, suggesting a negligible role of α7 nAChRs in nicotine-induced hyperlocomotion and reward in the rat [106]. Furthermore, it has been reported that chronic administration of both nicotine and SIB1765F, but not AR-R17779, resulted in an enhanced locomotor response to acute challenge with either nicotine or SIB1765F but not AR-R17779, suggesting that the α4β2 subtype plays a role in both the initiation and expression of sensitization to the psychomotor stimulant effects of nicotine [107]. Moreover, administration of AR-R17779 improved learning in two radial-arm maze tasks and reversed working memory impairment caused by fimbriafornix section [108]. These findings suggest that α7 nAChRs play a role in learning and memory, and that agonists of α7 nAChRs might have therapeutic potential for cognitive impairments in neuropsychiatric diseases, including schizophrenia.
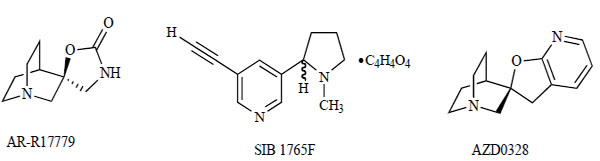
Chemical structures of AR-R17779, SIB1765F, and AZD0328.
Very recently, researchers at AstraZeneca reported that a furopyridine, (2'R)-spiro-[1-azabicyclo[2.2.2]octane-3,2'(3'H)-furo[2,3-b]pyridine d-tartrate (AZD0328; Fig. 4), was a novel selective partial agonist for an α7 nAChR [109]. AZD0328 exhibited high affinity for the native rat (Ki=4.7 nM) and recombinant human (Ki=3.0 nM) α7 nAChR, respectively. ADZ0328 also exhibited high affinity for both native rat (Ki=25 nM) and recombinant human (Ki=12 nM) 5-HT3 receptors. In contrast, AZD0328 exhibited only moderate affinity for the α4β2 nAChR of the native rat (Ki=140 nM) and very low affinity for the “ganglionic” α3 subunit-containing (Ki=2,500 nM) and muscle α1β1γδ nAChR (Ki=20,000) of the mouse. Functional assay revealed the partial agonistic nature of AZD0328. The half maximal (50%) effective concentration (EC50) and intrinsic activity of AZD0328 were 150 nM and 61% for rat α7 nAChR and 338 nM and 65% for human α7 nAChR, respectively. Administration of a low dose (0.00138 mg/kg) of AZD0328 in vivo leads to a significant increase in the excitability of midbrain dopaminergic neurons with no independent change in the spike patterns produced by these neurons. Within the same dose range, AZD0328 led to a significant increase in cortical dopamine release and improved both conditioned response learning and memory retention in an object recognition task. These effects were blocked by the selective α7 nAChR antagonist MLA (Fig. 2). AZD0328 improved novel object recognition in mice over a broad range of doses (0.00178–1.78 mg/kg) and the compound effect was found to be absent in homozygous α7 nAChR-knockout mice. Together, these data indicate that selective interaction with α7 nAChRs by AZD0328 selectively enhances midbrain dopaminergic neuronal activity, causing an enhancement of cortical dopamine levels; these neurochemical changes, in turn, likely underlie the positive behavioral responses observed in the two different animal models. These results also suggest that selective α7 nAChR agonists may have significant therapeutic utility in neurologic and psychiatric disorders in which cognitive effects and dopamine neuron dysfunction co-exist.
Mitsubishi Tanabe
Macor and Wu [110] reported some derivatives of 1-azabicyclo[2.2.2]oct-3-yl phenylcarbamate as agonists of α7 nAChRs (Fig. 5). In order to develop novel agonists of α7 nAChRs, researchers at Mitsubishi Pharma Corporation (currently Mitsubishi Tanabe Pharma Corporation) hypothesized that 1-azabicyclo[2.2.2]octane derivatives, bearing an aromatic part and a spacer group at the 3-position, may exhibit agonist activity at α7 nAChRs [111]. By examining a series of 3-substituted 1-azabicyclo[2.2.2]octane derivatives, they found that (+)-3-[2-(benzo[b]thiophen-2-yl)-2-oxoethyl]-1- azabicycle[2.2.2]octane was a potent and partial agonist of α7 nAChRs (Fig. 5) [111]. Furthermore, they reported the structure-activity relationships and pharmacokinetic profiles of the series of compounds leading to the discovery of (R)-3'(5-chlorothiophen-2-yl)spiro-1-azabicyclo[2,2,2]octane-3,5'-[1',3']-oxazolidin-2'-one (Fig. 5) [112]. This compound has potent binding affinity (Ki=9 nM for α7 nAChRs) and good selectivity toward the other nicotinic subtypes (α4β2 and α1β2γδ). Also, this compound has good oral bioavailability and brain permeability. Interestingly, this compound (10 mg/kg, p.o.) significantly improved dizocilpine (3 mg/kg)-induced auditory gating deficits in rats, suggesting that this compound has the potential to improve sensory gating deficits in schizophrenic patients [112]. Moreover, they have developed a novel partial α7 nAChR agonist, W-56203, (R)-3'-(3-methylbenzo-[b]thiophen-5-yl)spiro[1-azabicyclo[2.2.2]octane-3,5'-oxazolidin]-2'-one (Fig. 5) [113]. W-56203 bound to α7 nAChRs with a Ki value of 3 nM. No significant binding of W-56203 was detected at α4β2 nAChRs or muscarinic receptors. Furthermore, W-56203 showed no binding to other known receptors (dopamine D1 and D2, 5-HT1A, 5-HT2, adrenergic α1, α2, histamine H1 and H2) or ion channels (NMDA and α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA)), although W-56203 exhibited a moderate affinity to 5-HT3 receptors. In cultured hippocampal neurons, W-56203 evoked a rapidly desensitizing inward current, which was blocked by the selective α7 nAChR antagonist MLA (1 nM) (Fig. 2). Interestingly, W-56203 has been shown to significantly improve dizocilpine-induced auditory gating deficits in rats [113]. These results suggest that W-56203 is an orally active and partial agonist of α7 nAChRs, and that W-56203 would be a potential drug for the treatment of schizophrenia.
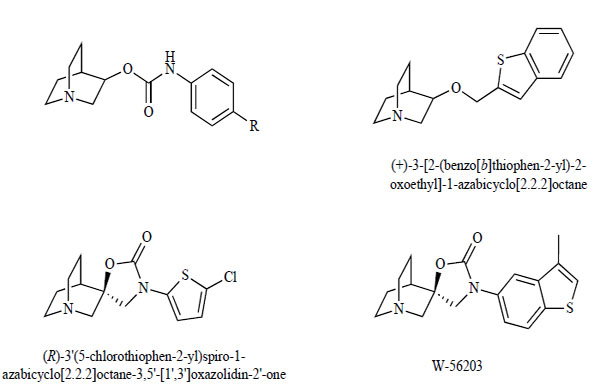
Chemical structures of 1-Azabicyclo[2.2.2]oct-3-yl phenylcarbamate derivatives, (+)-3-[2-(benzo[b]thiophen-2-yl)-2-oxoethyl]-1- azabicycle[2.2.2]octane, (R)-3’(5-chlorothiophen-2-yl)spiro-1-azabicyclo[2,2,2]octane-3,5’-[1’,3’]oxazolidin-2’-one, and W-56203.
Targacept
Researchers at Targacept Inc. reported that 2-(3-pyridyl)-1-azabicyclo[3.2.2]nonane (TC-1698; Fig. 6) was a highly selective agonist of α7 nAChRs [114]. TC-1698 exhibited a Ki of 11 nM in the binding assay of [3H]MLA to rat hippocampal membranes, whereas TC-1698 (10 μM) had no or very low affinity for other receptors. TC-1698 exerts neuroprotective effects via activation of the JAK2/PI3K cascade, which can be neutralized through activation of the angiotensin II receptors [114]. These findings suggest that JAK2 plays a central role in the α7 nAChR activation of the JAK2-PI3K cascade in PC12 cells, which ultimately contribute to α7 nAChR-mediated neuroprotection.
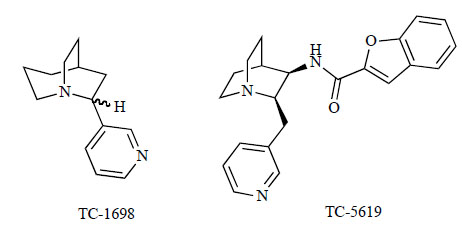
Chemical structures of TC-1698 and TC-5619.
They further synthesized a series of 2-(arylmethyl)-3-substituted quinuclidines as α7 nAChR agonists based on a putative pharmacophore model [115]. Quinuclidine is a well established pharmacophoric element. Its basic nitrogen, occupying a bridgehead position within an azabicyclic system, allocates the maximal electrostatic interaction combined with minimal steric demand. To further define pharmacophoric elements, they conducted virtual and synthetic diversification around a combination of 2-position and 3-position substitutions of the quinuclidine scaffold. This effort revealed the presence of three pharmacophoric elements: the cationic site (quinuclidine nitrogen), a hydrogen bond acceptor at the 3-position, and another hydrogen bond acceptor at the 2-position (the pyridine ring). In position 2 of quinuclidine, heteroarylmethyl groups are favored over benzyl, and steric constraints restrict substitution on the heteroaryl (pyridine) ring. The pharmacophoric element in position 3 was most amenable to diversification, resulting in the generation of four classes of ligands: carbamates, ureas, amides and sulfonamide. Cis-configuration is favorable for binding to the receptor. Within the 3-position substituent, a comparison of aliphatic and aromatic groups suggested that π-interaction may play a significant role in increasing affinity. As a result, several synthesized compounds exhibited high affinity to the α7 nAChR with Ki values near or below 1 nM. None of these compounds bound to α4β2 receptors with any significant affinity (Ki>10 μM). Among these series of compounds, N-{(2R,3R)-2-[(pyridin-3-yl)methyl]quinuclidin-3-yl}benzofuran-2-carboxamide (TC-5619; Fig. 6) has potent full agonistic activity for the α7 nAChR (EC50=33 nM, agonist activity=100), at a concentration below those that result in desensitization. Hauser et al. [116] further characterized the pharmacological properties of TC-5619. TC-5619 binds with very high-affinity to the α7 nAChR of the native rat hippocampus (Ki=1 nM) and the α7 nAChR of human HEK α7/RIC3 cells (Ki=1 nM), respectively. TC-5619 has little or no activity at other nicotinic receptors, including the α4β2 (Ki=2,800 and 2,100 nM for rat cortex and human SH-EP1 cells, respectively), ganglionic (α3β4) and muscle subtypes. In a transgenic th(tk–)/th(tk–) mouse model that reflects many of the developmental, anatomical, and multi-transmitter biochemical aspects of schizophrenia, TC-5619 acted both alone and synergistically with the antipsychotic clozapine to correct impaired pre-pulse inhibition (PPI) and social behaviors which model positive and negative symptoms, respectively. Similar to the results in the transgenic mice, TC-5619 significantly reversed the apomorphine-induced PPI deficits in rats. In a novel object-recognition paradigm in rats, TC-5619 demonstrated long-lasting enhancement of memory over a wide dose range. These findings suggest that TC-5619, either alone or in combination with antipsychotics, could offer a new approach to treating the constellation of symptoms associated with schizophrenia, including cognitive dysfunction. A phase 1 single rising dose (10–900 mg) clinical trial of TC-5619 in 66 healthy volunteers showed that TC-5619 was generally well tolerated up to doses of 600 mg*. A multiple dose (10–300 mg) administered for 10 days in 38 young healthy volunteers achieved significant improvements in the clinical dementia rating (CDR) test. A phase 2 proof-of-concept study will start at 4Q 2009 in patients with AD and attention deficit hyperactivity disorder (ADHD).
Pfizer
Researchers at Pfizer reported on the selective α7 nAChR agonist PNU-282987, N-[(3R)-1-azabicyclo[2.2.2]-oct-3-yl]-4-chlorobenzamide hydrochloride (Fig. 7) [117, 118]. PNU-282987 binds to α7 nAChR with a Ki of 27 nM, and showed evoked whole-cell currents from cultured rat hippocampal neurons that were sensitive to the selective α7 nAChR antagonist MLA [117, 118]. Systemic administration of PNU-282987 (1 mg/kg, i.v.) significantly improved d-amphetamine-induced sensory gating deficits in chloral hydrate-anesthetized rats. These findings suggest that PNU-282987 may be useful for treating the cognitive and attentional deficits of schizophrenia [117, 118]. This compound was later found, however, to possess significant human ether-a-go-go (hERG) potassium channel activity and thus did not meet Pfizer’s criteria for further development. Additional work on this template demonstrated that fused 6,5-heterocyclic analogues, such as indole (Fig. 7), provided an avenue toward novel analogues with potential for improved safety profiles. Of the 6,5-fused heterocycles evaluated, N-[(3R)-1-azabicyclo[2.2.2]oct-3-yl]furo[2,3-c]pyridine-5-carboxamide (PHA-543613; Fig. 7) proved to be a potent, high-affinity agonist of the α7 nAChR (Ki=8.8 nM: α7-5-HT3 chimera EC50=65 nM) [119]. PHA-543613 possesses antagonist activity at the 5-HT3 receptor (Ki=628 nM). PHA-543613 showed no detectable agonist activity (>100 μM) and negligible antagonist activity at both muscle-like nAChRs and ganglion-like nAChRs. Further, this furopyridine derivative did not significantly displace tritiated cytisine from rat brain homogenate at 1 μM, suggesting a selectivity over the α4β2 nAChR. The excellent in vitro profile of PHA-543613 is matched by rapid brain penetration, high oral bioavailability in rats, and a favorable hERG profile. Furthermore, PHA-543613 demonstrated efficacy in two in vivo models, the reversal of an amphetamine-induced P50 gating deficit (0.3 mg/kg, i.v.), and improved performance in a novel object-recognition test (1.0 mg/kg, s.c.).
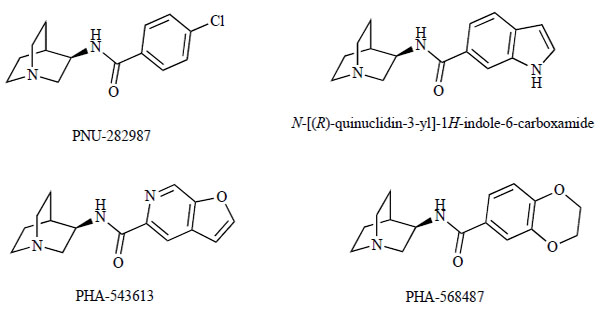
Chemical structures of PNU-282987, N-[(R)-quinuclidin-3-yl]-1H-indole-6-carboxamide, PHA-543613, and PHA-568487.
While further evaluation of PHA-543613 was underway, researchers at Pfizer identified additional novel, potent (<50 nM), and selective α7 nAChR agonists that, like PHA-543613, possess reduced hERG activity and low first pass metabolism [120]. To expand the SAR on the quinuclidine amide template and gain a broader understanding of the overlap with other nicotinic pharmacophores, two parallel approaches were utilized. The first focused on the acid portion of the quinuclidine template (amine part). The second approach investigated modifications to the azabicyclic ring system (acid part). The best compounds from each series are characterized by rapid brain penetration, good oral bioavailability in rats, and in vivo efficacy in a rat P50 auditory sensory gating assay. Finally, the researchhers discovered 2,3-dihydro-N-[(R)-quinuclidin-3-yl]benzo[b]-[1,4]dioxine-6-carboxamide (PHA-568487; Fig. 7), which showed an improved hERG safety profile over PHA-543613. However, these two phase, I clinical candidates (PHA-543613 and PHA-568487) were discontinued due to cardiovascular findings.†
Very recently, researchers at Pfizer disclosed the synthesis of (3R, 5R)-1-azabicyclo[3.2.1]octane-3-amine dihydrochloride (Fig. 8) and demonstrated that the activity of the corresponding para-chlorobenzamide (Fig. 8) in a α7-5-HT3 chimera assay was equal to that of the potent α7 nAChR agonist, PNU-282987. To identify novel, potent and orally bioavailable α7 nAChR agonists with in vitro and in vivo potential equal or better than that of PHA-543613, they proceeded to synthesis of an expanded set of amides derived from amine [121]. Among them, the furopyridine derivative N-[(3R,5R)-1-aza-bicyclo[3.2.1]octan-3-yl]furo[2,3-c]pyridine-5-carboxamide (PHA-709829; Fig. 8), which is a structurally similar analog of PHA-543613, showed very similar activity in the α7-5-HT3 chimera functional assay (EC50=81 nM) and similar affinity in the α7 binding assay (Ki=2.9 and 3.9). PHA-709829 showed stability in rat liver microsomes comparable to that of PHA-543613. PHA-709829 showed excellent CNS penetration in a brain delivery assessment screen (MDCK cell permeability, mouse brain/plasma ratio, Pgp substrate), displaying a profile very similar to PHA-543613.
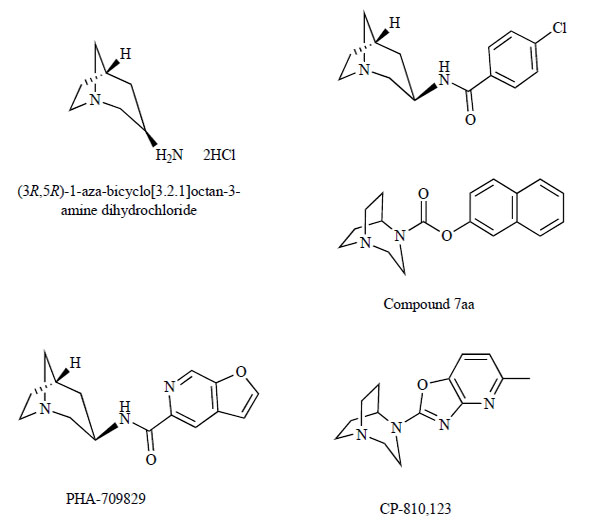
Chemical structures of (3R,5R)-1-aza-bicyclo[3.2.1]octan-3-amine dihydrochloride, para-chlorobenzamide derivatives of (3R,5R)-1-aza-bicyclo[3.2.1]octan-3-amine, PHA-709829, compound 7aa, and CP-801,123.
Researchers at Pfizer also identified 1,4-diazabicyclo[3.2.2]nonane phenyl carbamate (compound 7aa; Fig. 8) as subtype selective, high affinity α7 agonist (Ki=23 nM, agonist activity =175%). The pharmacological profiles of this compound closely resembled that of 4-bromophenyl 1,4-diazabicyclo[3.2.2]nonane-4-carboxylate hydrochloride (SSR180711; Fig. 9). The advantage of compound 7aa was that it showed the potential for an improved cardiosafety profile due to its low affinity for hERG.
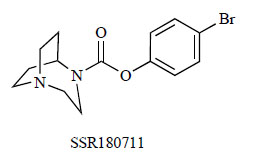
Chemical structure of SSR180711.
Very recently, they reported the benzoxyazole derivative 2-(5-methyloxazolo[4,5-b]pyridin-2-yl)-2,5-diaza-bicyclo[3. 2.2]nonane (CP-810,123; Fig. 8) as a novel α7 nAChR agonist.‡ This compound showed low affinity for hERG (IC50=40,000 nM) and high affinity binding to α7 nAChR. CP-810,123 has an excellent brain penetration and slow clearance from plasma and has shown marked ex vivo binding to cerebral binding sites (ED50=0.34 mg/kg). CP-810,123 also showed favorable preclinical findings in a number of animal models of cognition and was therefore advanced into clinical trials. However, the multiple-dose, 14-day clinical trial of CP-810,123 did not achieve any improvement of cognition at any of the doses tested, and was discontinued due to the occurrence of non-sustained ventricular tachycardia.
Sanofi-Aventis
Researchers at Sanofi-Aventis demonstrated a novel α7 nAChR agonist, SSR180711, 4-bromophenyl 1,4-diazabicyclo[3.2.2]nonane-4-carboxylate hydrochloride (Fig. 9) [122]. Binding assays show that SSR180711 has a high and selective affinity for the human (Ki=14 nM) and rat (Ki=22 nM) α7 nAChRs. This ligand inhibits, in a dose-dependent manner, the ex vivo [3H]α-bungarotoxin binding in mouse cortical homogenates after both i.p. and p.o. administration. At recombinant human α7 nAChRs, SSR180711 displays a partial agonist profile. Furthermore, it has been reported that SSR180711 improves cognitive deficits in a variety of rat models related to schizophrenia [123]. This drug restores the selective attention deficit induced by phencyclidine (PCP) administration at the neonatal stage. This action is reversed by the α7 nAChR antagonist MLA. This drug also restores a short-term episodic memory impairment and a spatial working memory deficit induced by PCP or dizocilpine.
Hashimoto et al. [124] reported that repeated administration of PCP significantly decreased the density of α7 nAChRs in the mouse brain. In addition, Hashimoto et al. [124] reported that SSR180711 could ameliorate cognitive deficits in mice after repeated administration of PCP, and that these effects could be blocked by the co-administration of MLA. Furthermore, Thomsen et al. [125] reported that repeated co-administration of SSR180711 (3 mg/kg) with PCP prevented both changes of parvalbumin and synaptophysin, which correspond to changes seen in patients with schizophrenia, and changes in the level of Arc, a molecule involved in synaptic plasticity, mRNA expression in the prefrontal cortex, and the behavioral impairment induced by PCP. Moreover, Barak et al. [126] reported that SSR180711 potentiated latent inhibition (LI) in normal rats and reversed amphetamine-induced LI disruption, two models considered predictive of activity against positive symptoms of schizophrenia [127, 128]. It was also reported that increase of expression of immediate early genes in the frontal cortex and nucleus accumbens has been demonstrated after administration with SSR180711 [129, 130]. Søderman et al. [131] reported that acute systemic administration of SSR180711 (10 mg/kg) resulted in a significant increase in Fos protein levels in the shell of the nucleus accumbens in wild-type mice, but had no effect in the Aβ1-42 overexpressing transgenic mice. This result suggests that overexpression of human Aβ peptides inhibits α7 nAChR-dependent neurotransmission in vivo, perhaps via direct interaction with α7 nAChR, and underscores that clinical trials testing α7 nAChR agonists should be related to the content of Aβ peptides in the patient’s nervous system.
Taken together, these findings suggest that SSR180711 has the potential to improve cognitive deficits associated with schizophrenia and AD.
WYETH/SIENA BIOTECH
Based on an initial hit identified in a screening program, researchers at Wyeth Research and Siena Biotech SpA obtained a novel α7 nicotinic acetylcholine receptor agonist, 5-morpholino-4-yl-pentanoic acid (4-pyridn-3-yl-phenyl)-amide (SEN12333/WAY-317538; Fig. 10), as the result of a structure-activity relationship study [132]. SEN12333/WAY-317538 shows high affinity for the rat α7 receptor expressed in GH4C1 cells (Ki=260 nM) and acts as a full agonist in functional Ca2+ flux studies (EC50=1.6 μM). SEN12333/WAY-317538 did not show agonist activity at the other nicotinic receptor subtypes tested (α1 and α4β2) or at highly homologous receptors (5-HT3A). SEN12333/WAY-317538 acted as a weak antagonist at α3-containing receptors and histamine H3 receptors. At a concentration of 10 μM of SEN12333/WAY-317538, no detectable inhibition binding was measured for the neurotransmitter and bioactive peptide receptors. SEN12333/WAY-317538 also showed minimal hERG inhibition, and desirable drug-like (molecular weight, number of rotatable bonds, log P, polar surface area) and pharmacokinetic properties. In vivo, SEN12333/WAY-317538 treatment (3 mg/kg i.p.) improved episodic memory in a novel object-recognition task in rats under the conditions of spontaneous forgetting and cognitive disruptions induced via glutamatergic (dizocilpine) or cholinergic (scopolamine) mechanisms [133]. This improvement was blocked by the α7 nAChR antagonist MLA. SEN12333/WAY-317538 also prevented a scopolamine-induced deficit in a passive avoidance task. In models targeting other cognitive domains, including attention and perceptual processing, SEN-12333/WAY-317538 normalized the apomorphine-induced deficit of PPI. Neuroprotection of SEN12333/WAY-317538 was demonstrated in quisqualate-lesioned animals in which treatment with SEN12333/WAY-317538 (3 mg/kg/day i.p.) resulted in a significant protection of choline acetyltransferase-positive neurons in the lesioned hemisphere. Cumulatively, these results demonstrate that the novel α7 nAChR agonist SEN12333/WAY-317538 has precognitive and neuroprotective properties.

Chemical structures of SEN12333/WAY-317538 and WYE-103914/SEN34625.
Very recently, researchers at Wyeth reported the novel selective α7 nAChR agonist 1-[6-(4-fluorophenyl)pyridin-3-yl]-3-[4-(piperidin-1-yl)butyl]urea (WYE-103914/SEN34625; Fig. 10).§ WYE-103914/SEN34625 bound with high affinity to a rat α7 nAChR expressed in GH4C1 cells with a Ki of 44 nM and increased intracellular Ca2+ with an EC50 of 130 nM (activity=99%). WYE-103914/SEN34625 was >100-fold selective against α1, α3, α4/β2 nAChRs receptors and the 5-HT3 receptor. In a rat novel object-recognition procedure, WYE-103914/SEN34625 (1 mg/kg, p.o.) treatment produced statistically significant enhancement of visual learning and memory retention, and this effect was blocked by pretreatment with MLA (5 mg/kg, i.p.). Pretreatment with WYE-103914/SEN34625 reversed the memory disrupting effects of dizocilpine (10 mg/kg, p.o.) in rats. WYE-103914/SEN34625, administered concurrently with dizocilpine immediately after training, produced a statistically significant reversal of dizocilpine-disrupted social odor recognition in mice at 3 mg/kg, p.o. In addition, WYE-103914/SEN34625 administered immediately following training enhanced retention memory in social odor recognition evaluated 6 days later (10 mg/kg, p.o.). Treatment with 3 mg/kg/day i.p. of WYE-103914/SEN34625 for 7 days also attenuated the decrease in the number of choline acetyltransferase-positive neurons produced by the injection of quisqualic acid into the nucleus basalis magnocellularis. These data indicate that WYE-103914/ SEN34625 exhibits a robust preclinical cognitive-enhancing and potentially neuroprotective profile.
Bayer
Researchers at Bayer reported the profile of the novel α7 nAChR agonist N-[(3R)-1-azabicyclo[2.2.2]oct-3-yl]-7-[2-(methoxy)phenyl]-1-benzofuran-2-carboxamide (ABBF; Fig. 11) [134]. ABBF bound to α7 nAChR in rat brain membranes (Ki=62 nM) and to recombinant 5-HT3 receptors (Ki=60 nM), and thus its agonistic activity was approximately 50-fold more potent than that of the natural agonist ACh (Ki=3 μM) and 10-fold more potent than nicotine (Ki=770 nM). ABBF was a potent agonist at the recombinant rat and human α7 nAChR expressed in Xenopus oocytes, but it did not show agonist activity at other nAChR subtypes. ABBF acted as an antagonist of the 5-HT3 receptor and α3β4, α4β2, and muscle nAChRs. ABBF improved social recognition memory in rats (0.3–1 mg/kg, p.o.). This improvement was blocked by intracerebroventricular administration of the α7 nAChR antagonist MLA (10 μg). In addition, ABBF improved the working memory of aged rats in a water maze repeated acquisition paradigm (1 mg/kg, p.o.) and object-recognition memory in mice (0.3–1 mg/kg, p.o.). Moreover, rats trained to discriminate nicotine (0.4 mg/kg, s.c.) from vehicle did not generalize to ABBF (0.3–30 mg/kg, p.o.), suggesting that the nicotine cue is not mediated by the α7 nAChRs and that selectiveα7 nAChR agonists may not share the abuse liability of nicotine. These results support the hypothesis that α7 nAChR agonists might provide a novel therapeutic strategy for the treatment of cognitive deficits with low abuse potential.
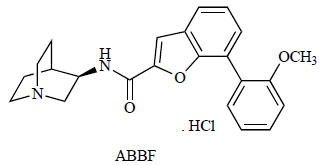
Chemical structure of ABBF.
Abott Laboratories
As mentioned above, the most recently described α7 agonists are derived from the quinuclidine structural class. Alternatively, researchers at Abott Laboratories identified 2,7-bis(2-(diethylamino)ethoxy)-9H-fluoren-9-one (Tilorone; Fig. 12) as a lead structure for a novel α7-selective agonist and developed related tricyclic analogs [135]. The lead structure tilorone bound α7 nAChR (IC50=110 nM) with high selectivity relative to α4β2 (IC50=70,000 nM), activated human α7 nAChR with an EC50 value of 2.5 μM and maximal response of 67% relative to ACh, and showed little agonist effect at human α3β4 or α4β2 nAChRs. However, the rat α7 nAChR maximal response was only 34%. Lead optimization led to 2-(5-methyl-hexahydro-pyrrolo[3,4-c]pyrrol-2-yl)-xanthen-9-one (A-844606; Fig. 12) with improved binding (α7: IC50=11 nM; α4β2: IC50>30,000 nM) and activity at both human and rat α7 nAChRs (EC50s=1.4 and 2.2 μM, respectively; apparent efficacies=61% and 63%). These compounds also activated native α7 nAChR, stimulating extracellular signal-regulated kinase 1/2 (ERK1/2) phosphorylation in PC12 cells. These findings suggest that tilorone, known as an interferon inducer, is a selective α7 nAChR agonist, and that the fluorene pharmacophore would be useful for the development of selective α7 nAChR agonists. Whether α7 nAChR stimulation mediates interferon induction, or whether interferon induction may influence the potential anti-inflammatory properties of α7 nAChR agonists remains to be elucidated.
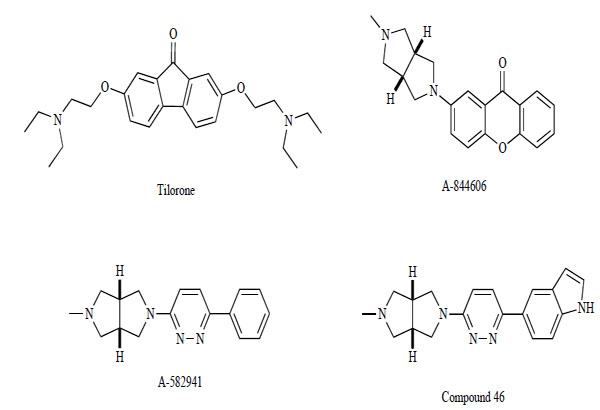
Chemical structure of Tilorone, A-844606, A-582941, and compound 46.
Researchers at Abbott Laboratories also reported a novel biaryl diamine α7 nAChR agonist, 2-methyl-5-(6-phenylpyridazin-3-yl)octahydropyrrolo[3,4-c]pyrrole (A-582941; Fig. 12). A-582941 was found to exhibit high-affinity binding to native rat (Ki=10.8 nM) and human (Ki=16.7 nM) α7 nAChRs [136, 137]. In contrast, A-582941 showed substantially weaker binding (Ki>100 μM) to α4, α3, and α1 subunit-containing nAChRs, respectively. Furthermore, A-582941 (10 μM) did not show significant displacement of binding of >75 targets (multiple G-protein-coupled receptors, ligand- and voltage-gated ion channel binding sites, acetylcholine esterase, and muscarinic receptors) with the exception of 5-HT3 receptors (Ki=154 nM). A-582941 showed partial agonism at Xenopus oocytes expressing human α7 nAChRs (EC50=4.26 μM; activity=52%) [137]. A-582941 showed acceptable pharmacokinetic properties and excellent distribution to the CNS in rodents. For example, intraperitoneal (i.p.) administration of A-582941 (1 μmol/kg) in mice produced maximal levels in the brain (286 ng/g at 0.33 h) that were 11-fold higher than the maximum plasma concentration (26 ng/mL at 0.25 h). This distribution ratio was maintained over an 8 h period, as the drug cleared from the brain and plasma with t1/2 = 2.5–3.2 h. Similarly, in rats, A-582941 distributes with approximately 6-fold higher levels in the brain than the plasma after i.p. dosing at 2-20 μmol/kg. dosing at 2–20 μmol/kg. Over this dose range, linear pharmacokinetics were observed, with the brain Cmax (normalized to a 1 μmol/kg dose) averaging 363 ng/g and maximum plasma levels of 62 ng/mL. A-582941 enhanced cognitive performance in behavioral assays20 μmol/kg. Over this dose range, linear pharmacokinetics were observed, with the brain Cmax (normalized to a 1 μmol/kg dose) averaging 363 ng/g and maximum plasma levels of 62 ng/mL. A-582941 enhanced cognitive performance in behavioral assays including the monkey delayed matching-to-sample, rat social recognition, and mouse inhibitory avoidance models, which capture domains of working memory, short-term recognition memory, and long-term memory consolidation, respectively. In addition, A-582941 normalized the sensory gating deficits induced by the α7 nAChR antagonist MLA in rats, and in DBA/2 mice that exhibit a natural sensory gating deficit. In vitro and in vivo studies indicated that A-582941 activates signaling pathways known to be involved in cognitive functions such as ERK1/2 and cAMP response element-binding protein (CREB) phosphorylation [137]. A-582941 exhibited a benign secondary pharmacodynamic and tolerability profile as assessed in a battery of assays of cardiovascular, gastrointestinal, and CNS function. A-582941 was non-mutagenic in the Ames reverse mutation assay in 5 bacterial strains, even with metabolic activation by a microsomal S9 fraction. The predominant adverse effects of A-582941, observed at doses substantially greater than those shown to be effective in cognition models, include clinical signs of CNS activity (tremors). Emesis and cardiovascular effects are of relatively minor concern. Although moderate QTc prolongation is observed with A-582941 in dogs at high plasma exposures, it does not appear to be α7-related. Investigation of potential carcinogenic effects produced no significant findings to suggest any α7-related toxicity. The details of the preclinical validation studies of A-582941 were recently reported [138]. Very recently, Biter et al. [139] reported that A-582941 can lead to increased phosphorylation of the inhibitory regulating amino acid residue Ser-9 on the glycogen synthase kinase3β (GSK3β), a major kinase responsible for tau hyperphosphorylation in the neuropathology of tauopathies such as front-temporal dementia and AD. A-582941 increased Ser-9 phosphorylation of GSK3β in the mouse cingulate cortex and hippocampus, and this increase in phosphorylation was not observed in α7 nAChR-knockout mice. Moreover, A-582941 continuous infusion decreased the phosphorylation of tau in hippocampal CA3 mossy fibers and spinal motoneurons in a hypothermia-induced tau hyperphosphorylation mouse and AD double transgenic APP/tau mouse model, respectively.
Very recently, researchers at Abbott Laboratories reported a series of 5-(pyridine-3-yl)octahydropyrrolo[3,4-c]pyrroles as a diamine scaffold for constitution of either α4β2 or α7 selective nAChR ligands [140]. In that article, they identified the most potent (agonist activity=87%) and selective (Ki for (7=0.24 nM; >400,000-fold selective for α4β2) α7 ligand in this series as 5-{6-[(3aS,6aR)-hexahydro-5-methylpyrrolo[3,4-c]pyrrol-2(1H)-yl]pyridazin-3-yl}-1H-indole (compound 46; Fig. 12).
Novartis
Researchers at Novartis reported a novel selective α7 nAChR agonist, (S)-(1-azabicyclo[2.2.2]oct-3-yl)-carbamic acid (S)-1-(2-fluoro-phenyl)-ethyl ester (JN403; Fig. 13) [141]. JN403 displayed high affinity for human recombinant α7 nAChR (Ki=200 nM). Ki values were estimated from the original pKd data. JN403 showed lower affinity for two other recombinant nAChR subtypes, human α3β4 (Ki=6,309 nM) and human α4β2 (Ki=158,489 nM). JN403 also showed low affinity toward the native murine 5-HT3 receptor (Ki=12,589 nM). Further, JN403 showed selectivity over a wide range of neurotransmitter receptors, ion channels and transporters. A functional assay with Xenopus oocytes expressing human recombinant α7 nAChR showed the partial agonistic nature of JN403 (EC50=2.14 μM; agonist activity=55%).
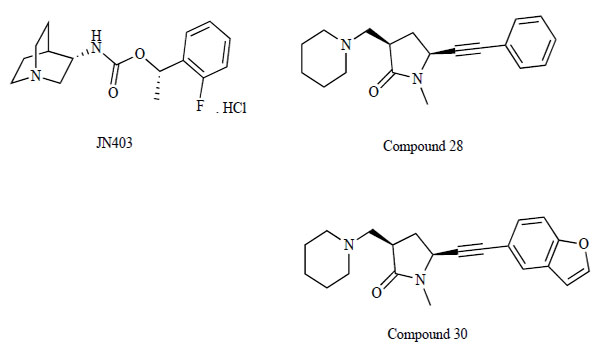
Chemical structure of JN403, compound 28, and compound 30.
Recently, in vivo effects of JN403 were reported by researchers at Novartis [142]. JN403 rapidly penetrates into the brain after intravenous (i.v.) and after peroral (p.o.) administration in mice and rats. In the social recognition test in mice JN403 facilitates learning/memory performance over a broad dose range. JN403 shows anxiolytic-like properties in the social exploration model in rats and the effects are retained after a 6 h pre-treatment period and after subchronic administration. Systemic administration of JN403 restores sensory gating in DBA/2 mice, a strain with reduced sensory inhibition under standard experimental conditions. Furthermore, JN403 shows anticonvulsant potential in the audiogenic seizure paradigm in DBA/2 mice. In the two models of permanent pain tested, JN403 produces a significant reversal of mechanical hyperalegesia. Together, these data suggest that JN403 may be beneficial for improving learning/memory performance, restoring sensory gating deficits, and alleviating pain, epileptic seizures and conditions of anxiety.
The common pharmacophore of nicotine and α7 nAChR agonists usually consists of a strong base (e.g., pyrolidine in nicotine; piperidine or tetrahydropyridine in an anabasein-derived scaffold; or a bicyclic amine, usually quinuclidine) connected to a hydrophilic element by a linker (e.g., carbamate, ether or amide). In view of the highly populated chemical space around these general scaffolds, researchers at Novartis discovered alternative templates. They found a cis γ-lactam scaffold and optimized it to reveal highly potent and selective α7 nAChR agonists with in vitro activity and selectivity and with good brain penetration in mice [143]. Among them, (3R,5S)-1-methyl-5-(2-phenylethynyl)-3-[(piperidin-1-yl)methyl]pyrrolidin-2-one (compound 28) and (3R,5S)-5-[2-(benzofuran-5-yl)ethynyl]-1-methyl-3-[(piperi-din-1-yl)methyl]pyrrolidin-2-one (compound 30) showed potent and selective α7 nAChR agonistic activity (Fig. 13). Compounds 28 and 30 displayed high affinity for human recombinant α7 nAChR (IC50=10 and 7.9 nM, respectively). The IC50 values were estimated from the original pIC50 data. At other recombinant nAChR subtypes, compounds 28 and 30 showed lower affinity: human α3β4 (IC50=1,258.9 and 1,000 nM, respectively); human α4β2 (IC50=7,943.3 and 10,000 nM, respectively); human α1β1γδ (IC50=12,589.3 and 6,309.6 nM, respectively). Compounds 28 and 30 also showed low affinity toward recombinant human 5-HT3 receptors (IC50=251,188.6 and 50,118.7 nM, respectively). When tested against human recombinant muscarinic receptors, compounds 28 and 30 also displayed an IC50>10 μM on all subtypes (M1-M5). Compound 28 showed rapid clearance, but the brain/plasma ratios remained high at 1 and 4 h after treatment with 30 μmol/kg p.o.. Compound 30 improved the in vivo pharmacokinetic properties. High brain and plasma levels were observed at 1 and 4 h following administration. Additionally, these compounds displayed good cardiosafety profiles. Taken together, these findings indicate that these new scaffolds, which are devoid of the classical bicyclic amine, will provide a valuable lead series for further development.
Eli Lilly
Researchers at Eli Lilly found several high affinity ligands for α7 nAChRs with no/small 5-HT3 receptor cross-reactivity [144]. For example, N-(quinuclidin-3-yl)-5-(thiophen-2-yl)thiophene-2-carboxamide (compound 8; Fig. 14) showed a potent α7 ligand (Ki=1.1 nM) and still had 1,000-fold selectivity for α7 nAChRs over 5-HT3 receptors. Additionally, 5-phenyl-N-[(R)-quinuclidin-3-yl]thiophene-2-carboxamide (compound 7a; Fig. 14) also showed a potent α7 ligand (Ki=1.8 nM) and high selectivity for α7 nAChRs over 5-HT3 receptors (5-HT3 receptor Ki: 24% at 10 μM).
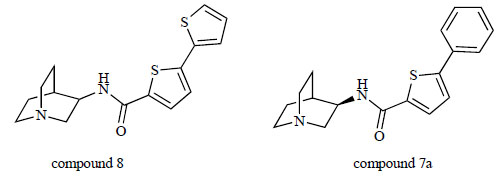
Chemical structure of compound 8 and compound 7a.
MEMORY PHARMACEUTICALS/ROCHE
Researchers at Memory Pharmaceuticals reported a novel α7 nAChR partial agonist and 5-HT3 receptor antagonist, R3487/MEM3454 [145]. The chemical structure of R3487/MEM3454 was not disclosed, and the compound is currently being developed by Roche as RO5313534 [146]. R3487/MEM3454 is an orally active α7 nAChR agonist with high affinity for native rat α7 nAChRs (Ki=6 nM). R3487/MEM3454 also showed a similar high affinity binding to human recombinant 5-HT3 receptors (Ki=2 nM). Functionally, R3487/MEM3454, with a molecular weight of 306.79, acted as a partial agonist in monkey recombinant α7 nAChRs (EC50=0.4 nM) and displayed an antagonist action against native guinea pig 5-HT3 receptors. R3487/MEM3454 lacked affinity at α4β2 nAChRs and had no activity at other nicotinic or other CNS receptor subtypes. Additionally, R3487/MEM3454 was demonstrated to be efficacious in several behavioral paradigms representing multiple cognitive domains (episodic, spatial, working, attentional and executive memory functions) in young and aged rodents as well as non-human primates.** Interestingly, R3487/MEM-3454 can increase both dopamine and ACh efflux in the rat medial prefrontal cortex and hippocampus when administered subcutaneously. These effects were completely blocked by MLA in both brain regions.
Currently RO5313534 (formerly R3487/MEM 3454) is being developed for treatment of AD and cognitive impairment in schizophrenia. Three different doses of RO5313534 (as R3487/MEM3454) were evaluated in a phase IIa trial in 80 patients with mild to moderate AD [146]. The 2 lower doses (5 and 15 mg/day) were associated with significant improvements compared with placebo on several efficacy measures (P≤0.05). Constipation was the only adverse event that was found to occur substantially more frequently in the R3487/MEM3454 groups compared with the placebo group (43% vs 5%, respectively); no serious adverse events were attributed to the study drug. A second phase II trial was initiated in April 2009 to assess the safety and efficacy of 4 different dosages of RO5313534 as an adjunctive therapy to donepezil 5 or 10 mg/day in patients with mild to moderate AD.
Servier
Researchers at Servier reported 2-[2-(4-bromophenyl)-2-oxoethyl]-1-methyl pyridinium, S 24795 (Fig. 15), as a partial agonist for α7 nAChRs [147]. S 24795 was selected from a series of bromophenyl pyridinium derivatives. S 24795 showed a moderate affinity to native rat α7 nAChR (IC50=4.6 μM) but did not show any affinity to α4β2 nAChR, muscle-type nicotinic receptors or ganglionic-type nicotinic receptors. S 24795 is a partial agonist of α7 nAChR with an EC50 of 34 ± 11 μM and approximately 10% activity relative to ACh. S 24795 enhances long-term potentiation at CA3-CA1 synapses in the adult mouse hippocampus [148]. This effect was considered to be mediated by α7 nAChRs, since it was prevented by MLA (10 μM) and was absent in α7-knockout mice. S 24795 improved contextual memory in aged mice [149] and aging-related deficits in declarative and working memory in mice [150]. Interestingly, in vitro exposure of S 24795 reduced the existing Aβ42–α7 nAChR complexes in AD frontal cortex synaptosomes [94]. Furthermore, in vitro exposure of S 24795 normalized α7 nAChR- and NMDA receptor-mediated Ca2+ influx and improved NMDA receptor signaling in AD and Aβ42-exposed control brain synaptosomes. S 24795 facilitates Aβ42 release from Aβ42–α7 nAChR and –Aβ42 complexes by interacting with the Aβ15-20 region of Aβ42, the pivotal Aβ42-binding domain to the α7 nAChR.
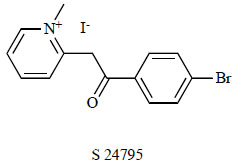
Chemical structure of S 24795.
EnVivo
Very recently, researchers at EnVivo Pharmaceuticals Inc. reported EVP-6124 as a novel α7 nAChR agonist.†† The chemical structure of EVP-6124 was not disclosed. EVP-6124 is a selective and potent α7 nAChR agonist (Ki = 4.3 nM) that has antagonist activity at the 5-HT3 receptor (IC50 = 299 nM), and limited or no activity on other receptors. EVP-6124 has an excellent brain to plasma ratio and has shown marked efficacy and potency in a number of animal models of cognition. In rats with scopolamine-induced memory deficit, a single dose of EVP-6124 reverses the short-term memory impairment in an object-recognition task. In a model of natural forgetting of the novel objective-recognition task, EVP-6124 was effective at preventing natural forgetting when given after the first trial. EVP-6124 was also effective in this model when given after the first trial or when given only before the second trial. Furthermore, EVP-6124 also reversed scopolamine-induced deficits in a water maze repeated acquisition task model. These observations suggest that EVP-6124 may enhance acquisition as well as consolidation and retrieval processes. Further preclinical investigation of EVP-6124 revealed a large safety margin relative to systemic exposure of EVP-6124 based on the lack of an observable adverse effect level in dogs. In phase 1 clinical trials, the safety and tolerability of EVP-6124 has been demonstrated in healthy volunteers as well as in subjects with schizophrenia and AD. The efficacy of EVP-6124 is currently being investigated in AD as well as in cognitive impairment associated with schizophrenia.
Tropisetron
Both α7 nAChRs and 5-HT3 receptors are members of the superfamily of ligand-gated ion channels [23, 28]. These two receptors share the greatest similarity within the family, displaying approximately 30% sequence homology [151]. Tropisetron and ondansetron (Fig. 16) are potent 5-HT3 receptor antagonists that are widely used in the treatment of patients with chemotherapy-induced or postoperative nausea and vomiting [23]. It has been reported that tropisetron is a partial agonist of α7 nAChRs with a high affinity, whereas ondansetron has a weak affinity at α7 nAChRs [152, 153]. We found that tropisetron improves the deficient inhibitory processing of P20-N40 in DBA/2 mice, and that improvement by tropisetron could be antagonized by co-administration of MLA [154]. Furthermore, we reported that PCP-induced cognitive deficits could be improved by subsequent subchronic administration of tropisetron, but not ondansetron, and that improvement by tropisetron could be antagonized by co-administration of MLA [155]. These findings suggest that tropisetron could improve abnormal auditory gating of P20-N40 and PCP-induced cognitive deficits in mice via α7 nAChRs. In addition, we reported that tropisetron (10 mg) could improve deficits of P50 suppression in schizophrenic patients [156]. A randomized, placebo-controlled study of tropisetron on cognitive dysfunction in schizophrenic patients is currently underway at Chiba University.

Chemical structure of tropisetron and ondansetron.
CONCLUDING REMARKS
As described above, accumulating evidence suggests that α7 nAChRs play an important role in the pathophysiology of neuropsychiatric diseases, including schizophrenia and AD. Hence, a number of pharmaceutical industries have developed selective and high affinity α7 nAChR agonists as therapeutic drugs for these neuropsychiatric diseases. Over the past decade, a variety of largely quinuclidine pharmacophore-based agonists of α7 nAChRs have emerged, including AR-R17779, PNU-282987, PHA-543613, PHA-568487, W-56203, ABBF and JN403. In the past 10 years, several α7 nAChR agonists (DMXB-A, PHA-543613, PHA-568487, R3487/MEM 3545 and tropisetron) have entered clinical trials for potential use in schizophrenia or AD. Today, lead optimization efforts and structure-activity relationship studies have identified several different pharmacophore-based compounds, including biarylamine derivatives (TC-1698, TC-5619, PHA-709829, SSR180711, A-844606 and A-582941) and others. Some of these new compounds have entered preclinical validation studies as candidates for clinical trial.
In vivo positron emission tomography (PET) imaging of α7 nAChRs in the intact human brain provides a method for quantitative study of α7 nAChR-related pathophysiology in neuropsychiatric diseases [157]. Very recently, we developed the novel PET ligand, [11C]CHIBA-1001 (Fig. 17), for α7 nAChRs in the human brain [158, 159]. To the best of our knowledge, there are no other PET ligands for α7 nAChRs available for human use. The in vivo determination of α7 nAChR receptor occupancy using PET and [11C]CHIBA-1001 allows for demonstration of the target engagement and assessment of the titration for potential dose regimens in humans. In addition, a clinical PET study in patients with schizophrenia or AD using [11C]CHIBA-1001 is currently underway.
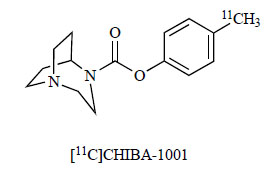
Chemical structure of [11C]CHIBA-1001.
NOTES
* Lippiello, P. TC-5619: An α 7 nAChR selective agonist with efficacy in animal models of positive and negative symptoms, and cognitive dysfunction of schizophrenia.
Nicotinic acetylcholine receptors as therapeutic targets: emerging frontiers in basic research and clinical science; Chicago, IL, Oct. 14-Oct. 17, 2009.
† Rogers, B.N. Agonists of α 7 nAChRs for the potential treatment of cognitive deficits in Schizophrenia. Nicotinic acetylcholine receptors as therapeutic targets: emerging frontiers in basic research and clinical sciences; San Diego, CA, Oct. 31-Nov. 2, 2007.
‡ O’Donnell, C.J. CP-810,123 a third generation alpha-7 nAChR agonist for treatment of cognitive deficits associated with schizophrenia. Nicotinic acetylcholine receptors as therapeutic targets: emerging frontiers in basic research and clinical science; Chicago, IL, Oct. 14-Oct. 17, 2009.
§ Dunlop, J.; Comery, T.A.; Lock, T.; Kramer, A.; Kowal, D.; Yeola, S.; Jow, F.; Aschmies, S.; Lin, Q.; Beyer, C.E.; Brennan, J.; Kelly, C.; Roncarati, R.; Scali, C.; Haydar, S.; Ghiron, C.; Marquis, K.L.; Harrison, B.; Robichaud, A.; Terstappen, G.C. In vitro pharmacological characterization and pro-cognitive effects of the selective alpha-7 nicotinic agonist WYE-103914. Biochem. Pharmacol., 2009, 78, 911.
** Wallace, T.L.; Chiu, H.; Dao, D.A.; Lowe, D.A.; Porter, R.; Santarelli, L. R3487/MEM 3454, a novel nicotinic α7 receptor partial agonist, improves attention and working memory performance in cynomolgus macaques. Biochem. Pharmacol., 2009, 78, 912.
†† Koenig, G. EVP-6124, a novel α 7 nAChR agonist for the treatment of cognitive impairments in Alzheimer’s disease & schizophrenia. Nicotinic acetylcholine receptors as therapeutic targets: emerging frontiers on basic research and clinical science; Chicago, IL, Oct. 14-Oct. 17, 2009.
ACKNOWLEDGEMENTS
This study was supported by a grant from the Program for Promotion of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation of Japan (to K.H., ID#:06-46).

