All published articles of this journal are available on ScienceDirect.

A Single Amino Acid Substitution Makes WNK4 Susceptible to SB 203580 and SB 202190
Abstract
Regulation of the SLC12 family of membrane transporters including NCCT involves a scaffold of interacting proteins including the STE 20 kinase SPAK and the WNK kinases, WNK 1 and WNK 4, which are mutated in the hypertensive syndrome of pseudohypoaldosteronism type 2 (PHAII). WNK4 regulates NCCT by affecting forward trafficking to the surface membrane. Studies in Xenopus using kinase dead WNK4 site mutants have produced inconsistent results with regard to the necessity of kinase function for NCCT regulation. Dynamic inhibition of WNK4 by small molecules may bring clarity to this issue however WNK4 is naturally resistant to commercial MAP kinase inhibitors owing to steric constraints prohibiting entry of small molecules to the active site. Using an approach similar to that used in p38 and ERK, we show that a single substitution in WNK4 (T261G) dramatically enhances its susceptibility to the inhibitors SB 202190 and SB 203580.
INTRODUCTION
Protein kinases form a fundamental network of intracellular signalling molecules necessary for most human signal transduction pathways. Abnormal kinase function has been associated with disease processes as diverse as cancer [1], diabetes [2], rheumatoid arthritis [3] and hypertension [4]. Recent therapeutic advances using small cell permeable kinase inhibitors have yielded dramatic results; for example the BCR-ABL inhibitor Imatinib, effective in Chronic Myeloid leukaemia [5] and the mTOR inhibitor Rapamycin for preventing transplant rejection [6] and have also revolutionised our understanding of signal transduction pathways themselves [7].
Kinases may contribute to hypertension either by augmenting arteriolar vasoconstriction by Rho kinase [8] or by increasing sodium retention in the distal renal tubule through regulation of ENaC by SGK1 [9] and NCCT by WNK kinases [4]. WNK kinases (With No Lysine Kinases) are a family of four kinases of which the genes for WNK1 and WNK4 are mutated in Gordon syndrome or Pseudohypoaldosteronism type II (PHA II), a rare monogenic form of hypertension. A model in which WNK1 inhibits WNK4 that in turn inhibits NCCT forward trafficking to the apical membrane is well established [4], although the effect of WNK4 kinase function in regulating NCCT expressed in Xenopus using kinase dead site mutants has yielded inconsistent results [10-12]. A major limitation in the exploration of WNK kinase function in Xenopus as well as in tissues and whole animals has been the absence of specific inhibitors.
The pyridinyl imidazoles SB 203580 and SB 202190 were developed at SmithKline Beecham as inhibitors of IL-1 and TNF production in activated monocytes [13]. Radioligand binding assay subsequently identified the kinase targets now known as the stress activated MAPK kinases, SAPK2a/p38α and SAPK2b/p38β. These SB compounds were used to elucidate some of the physiological effects of this pathway [14], as well as identifying downstream targets of the kinases [14-16]. X-ray crystallography of SB 203580 bound to p38 kinase revealed the drug binding to the ATP binding pocket and allowed adjacent residues in the pocket to be identified [17]. Threonine106 is located close to the 4-fluorophenyl ring of the inhibitor Fig. (1) and mutation to the more bulky M106 retards kinase inhibition [14, 17, 18]. In fact, a series of T106 p38 mutants showed a hierarchy of kinase inhibition with smaller residues such as glycine or alanine allowing maximum drug binding. Using this approach a previously insensitive kinase, ERK 2, was made drug-sensitive by mutation of the homologous residue Q105 to the smaller T105 [19]. Four other closely located residues were also found to modulate ERK2 sensitivity to SB 203580 in a complementary manner.
Chemical structure of the p38 kinase inhibitors SB202190 and SB 203580.
We hypothesised that in WNK4, mutation of the homologous amino acid T261 to glycine might facilitate binding of small molecule kinase inhibitors such as the SB compounds to the ATP pocket and so confer drug sensitivity permitting dynamic investigation of WNK4 kinase function on the regulation of NCCT.
MATERIALS AND METH0DOLOGY
Cloning and cRNA Synthesis
Wild type NCCT in ECFP-TNT with cyan fluorescent protein at its N terminal and wild type WNK4 in pcDNA3 were as previously described [11]. The WNK4 cDNA was mutated to T261G using site directed mutagenesis (Stratagene, La Jolla, CA). All sequences were verified using an ABI 377 and Big Dye fluorescent chemistry (Applied Biosystems, Foster City, CA). Copy RNA was transcribed in vitro from linearized plasmids using the T7 mMESSAGE mMACHINE® kit (Ambion, Austin, Tx) and quantified using ultraviolet absorption spectroscopy (Nanodrop, Wilmington, DE).
Expression in Xenopus Oocytes
Xenopus laevis oocytes were harvested and defolliculated as detailed previously [20]. Briefly, 10 ng of NCCT cRNA was injected in a total volume of 50nl per oocyte, and for coinjections involving WNK4 an additional 10ng cRNA was added to the injectate. RNAase and DNAase free water-injected oocytes were used as controls throughout. Oocytes were then incubated in ND96 containing 2 mM sodium pyruvate, 0.1mg/ml gentamicin and kinase or inhibitors at 18oC for 5 days.
For 22Na+ flux studies, oocytes were placed for 24 hours in Cl- free ND96 solution containing 96mM sodium isethionate, 2mM potassium gluconate, 1.8 mM calcium gluconate, 1mM magnesium gluconate, 5mM HEPES, 2.5mM sodium pyruvate and 5mg/dl gentamicin. Thirty minutes before the addition of uptake medium, oocytes were added to Cl- free ND96 with inhibitors (1mM ouabain, 100µM amiloride and 100µM bumetanide) according to the protocol of Gamba [21] . Oocytes were then transferred to isotonic uptake medium (58mM NaCl, 38mM M-methyl-D-glucamine, 2mM KCl, 1.8mM CaCl2 and 5mM HEPES with inhibitors pH 7.4) containing 22Na+ at a final concentration of 2.5µCi/ml and incubated in a gently shaking incubator at 37oC for 1 hour. Oocytes were then washed five times with 3ml ice cold aliquots of isotonic medium and the oocytes counted individually in a gamma counter (Perkin-Elmer Cobra 5003). Thiazide sensitivity was confirmed using 100µM hydrochlorthiazide (data not shown).
Membrane surface expression measurements were performed 5 days after injection by laser-scanning confocal microscopy with a Leica DMRXA confocal microscope. Data was captured using an equatorial section through each oocyte under a x10 objective lens with brightness, contrast and pin-hole settings kept constant for all oocytes in each injection series. The fluorescent signal in the membrane was quantified using Leica confocal software (version 2.61 of LCS Lite®) with sampling made at sixteen equispaced points on the circumference and averaged to give mean total fluorescence intensity in arbitrary fluorescence units (AFUs).
Data Analysis
For all oocyte experiments ten to fifteen oocytes were injected for each cRNA used. Differences between groups were compared by 1-way ANOVA with post hoc testing. Figures show representative experiments that were replicated using at least four different batches of oocytes from different donor animals. The SPSS statistical package (software version 12) was used throughout with significance defined as P < 0.05. The IC50 values for the SB compounds were derived from the fitting of the dose-response curves to a simple logistic function using GraphPad version 4.
RESULTS
T261G Mutant WNK4 Behaves as Wild Type WNK4
22Na+ flux measurable in NCCT injected oocytes was completely inhibited by hydrochlorothiazide (100 μM) as shown in Fig. (2A). Wild type WNK4 coinjection with NCCT decreased 22Na+ uptake by 62% (3.0 +/- 0.1 vs. 7.9 +/- 0.4 nmol/oocyte/h, P<10-4) in keeping with previous reports. T261G WNK4 produced a similar effect reducing 22Na+ uptake through NCCT by 71% (2.3 +/- 0.4 vs. 7.9 +/- 0.4 nmol/ oocyte/h, P< 10-4), which was not significantly different from wild type WNK4 (3.0 +/- 0.1 vs. 2.3 +/- 0.4 nmol/oocyte/h, P> 0.05).
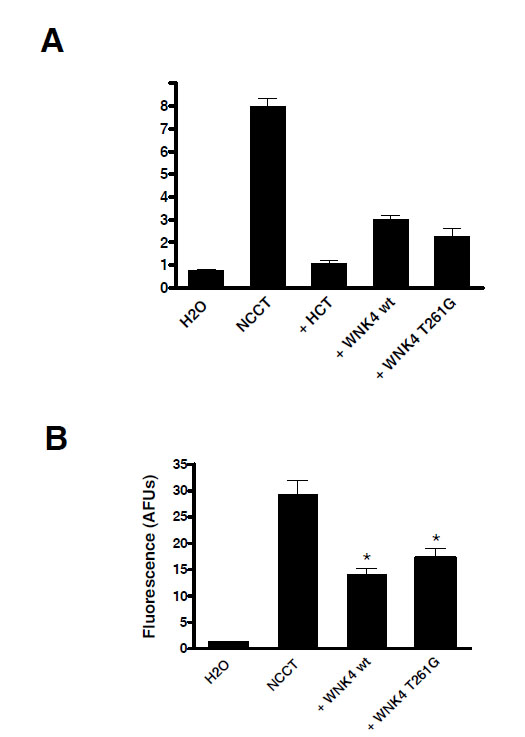
The T261G WNK4 mutant behaves like wild type WNK4. A. Oocytes were injected with NCCT wt alone or in combination with WNK4 wt or WNK4 T261G. Mutant WNK4 reduced 22Na+ flux equivalent to the same extent as WNK4 wt. * P< 0.0001vs NCCT alone. B. Confocal microscopy of the fluorescent signal from eCFP-NCCT showed that changes in WNK4 induced 22Na+ flux were accompanied by similar changes in surface membrane expression of the cotransporter. * P< 0.0001 vs NCCT alone.
Confocal microscopy of ECFP-NCCT injected oocytes showed that WNK4 effects on NCCT flux activity were accompanied by parallel changes in surface membrane expression of the transporter Fig. (2B). Wild type WNK4 decreased membrane expression of ECFP-NCCT by 52% (14.1 +/- 1.1 vs. 29.4 +/- 2.6 AFUs, P< 10-4). Similarly T261G WNK4 reduced NCCT surface membrane abundance by 41% (17.2 +/- 1.8 vs. 29.4 +/- 2.6 AFUs, P< 10-4). Again the effects of the two forms of WNK4 kinases were not significantly different from each other (14.1 +/- 1.1 vs. 17.2 +/- 1.8 AFU’s, P >0.05).
Site Mutation of WNK4 Confers Kinase Inhibitor Sensitivity
Xenopus oocytes were injected with cDNA for ECFP-NCCT with either wild type or the mutant WNK4 T261G. They were then incubated with low (0.5 µM) or high (5µM) concentrations of the SB compounds to see if these kinase inhibitors were able to block the catalytic activity of WNK4 and restore 22Na+ uptake. Fig. (3A) shows that 22Na+ uptake by wild type WNK4 injected oocytes was unaffected by incubation with either concentration of the SB inhibitors indicating that the IC50 for both SB compounds must be >5 µM against wild type WNK4. Conversely 22Na+ flux through NCCT coinjected with mutant WNK4 T261G was increased when incubated with either inhibitor; the flux actually returning to the level seen in oocytes injected with NCCT alone.
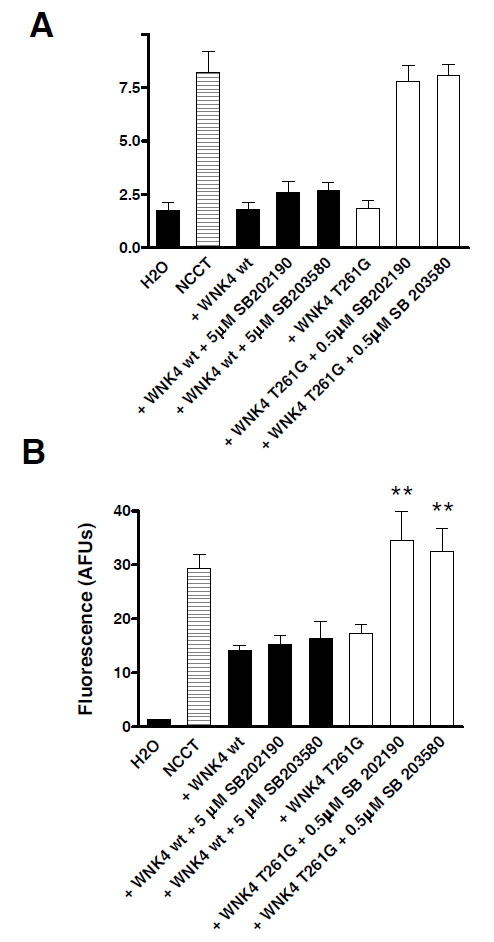
The T261G mutation of WNK4 confers kinase inhibitor sensitivity. A. Oocytes were injected with NCCT wt alone or in combination with either WNK4 wt or WNK4 T261G. The 22Na+ flux assay was then performed with or without 0.5 or 5 µM of each SB compound. * P<0.0001 vs NCCT alone. B. Confocal microscopy of the fluorescent signal from eCFP-NCCT showed that changes WNK4-induced 22Na+ flux were accompanied by parallel changes in surface membrane expression of the cotransporter. * P< 0.05 vs assay without an SB inhibitor.
The changes in 22Na+ flux, were accompanied by parallel changes in the membrane fluorescence and hence membrane expression of NCCT Fig. (3B).
Concentration Response Curves for SB 202190 and SB 203580
These results in Fig. (3) suggested that the IC50 for both SB compounds was at ~100-fold lower for the T261G mutant versus wild type. To confirm this, concentration-response curves were generated for the SB compounds in oocytes expressing WNK4 T216G. The fitted IC50 values were 109 nM for SB 203580 and 147 nM for SB 202190 Fig. (4).
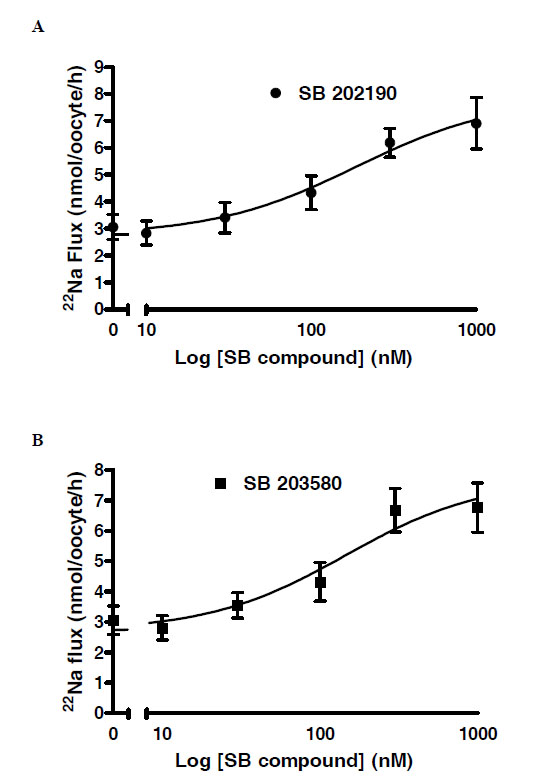
Concentration response curves showing reversal of WNK4 T261G inhibition of NCCT 22Na+ flux in Xenopus oocytes by (A) SB 202190 and (B) SB203580. The fitted EC50 values (concentration of inhibitor producing 50% reversal) were respectively 147 and 109 nM.
DISCUSSION AND CONCLUSION
Experiments to study the effects of WNK4 in whole animals and tissues have been hampered by the lack of an inhibitor. Although it is well established that wild type WNK4 inhibits NCCT forward trafficking to the surface membrane [4], it was unclear whether in tact kinase function was necessary for this to occur. Studies using kinase dead WNK4 site mutants expressed in Xenopus have yielded inconsistent results [10-12].
Within the kinome, the WNKs show closest homology to the MAPKK subgroup of MAPK kinases. Inhibitors of the MAPK kinase family are well described, although the sensitivity of individual kinases is dependent on steric factors around the ATP binding pocket. In keeping with reports using p38 and ERK2 kinases [14-16, 19], we found that mutation of the single threonine residue at position 261of WNK4 (homologous amino acid 106 in p38) to a residue with a much smaller side chain resulted in dramatic enhancement of its sensitivity to SB 202190 and SB203580. The IC50 for both agents shifted from >5000 nM to ~100 nM, so that both inhibitors were able to completely reverse the effect of WNK4 T261G on NCCT in the oocyte system. These results confirm that WNK4 requires a catalytically intact kinase function to affect NCCT trafficking [10, 11].
It is hoped that further study of the steric constraints preventing small molecules from gaining access to the active site of WNK kinases may enable the design of compounds capable of inhibiting wild type WNK kinases. The availability of small molecule inhibitors of wild type WNK4 may be attractive therapeutically. Salt-retention through increased expression of NCCT could be useful, for example, in hypotensive states such as autonomic failure. It is not clear that a similar strategy could be used for WNK1. Heterozygous mouse knock-outs for WNK1 do have a lowered blood pressure [22], but there are contradictory reports of the necessity for intact WNK1 kinase function to influence NCCT via WNK4 [11, 12]. WNK1 is also found predominantly in the short form which lacks the kinase domain [23], although in states of low potassium diet [24] or Gordon syndrome, the full length transcript may be physiologically more significant [25, 26].
In summary, we have shown that T261G mutation of WNK4 renders it susceptible to inhibition by SB202190 and SB203580 and confirming that the ability of WNK4 to regulate NCCT is indeed dependent on kinase activity. This may be useful in elucidating the role of WNK4 kinase activity in regulating renal electrolyte transport experimentally.
GRANTS
MG was supported by a British Heart Foundation (UK) Clinical Studentship FS/06/034 and a Raymond and Beverly Sackler Studentship.

