All published articles of this journal are available on ScienceDirect.

Screening of Potential anti-Trypanosoma cruzi Candidates: In Vitro and In Vivo Studies
Abstract
Chagas disease (CD), caused by the intracellular protozoan Trypanosoma cruzi, is a parasitic illness endemic in Latin America. In the centennial after CD discovery by Carlos Chagas (1909), although it still represents an important public health problem in these affected areas, the existing chemotherapy, based on benznidazole and nifurtimox (both introduced more than four decades ago), is far from being considered ideal due to substantial toxicity, variable effect on different parasite stocks and well-known poor activity on the chronic phase. CD is considered one of the major “neglected” diseases of the world, as commercial incentives are very limited to guarantee investments for developing and discovering novel drugs. In this context, our group has been pursuing, over the last years, the efficacy, selectivity, toxicity, cellular targets and mechanisms of action of new potential anti-T. cruzi candidates screened from an in-house compound library of different research groups in the area of medicinal chemistry. A brief review regarding these studies will be discussed, mainly related to the effect on T. cruzi of (i) diamidines and related compounds, (ii) natural naphthoquinone derivatives, and (iii) megazol derivatives.
1. INTRODUCTION
Chagas disease (CD) is a neglected illness caused by the protozoan Trypanosoma cruzi. Its life cycle involves haematophagous triatomine insects (vectors), diverse vertebrate mammalian hosts and different developmental forms of the parasite. Briefly, after the ingestion of bloodstream trypomastigotes by the insect vectors from an infected mammalian host, there is the conversion to epimastigote forms, which proliferate and then differentiate into metacyclic forms within the posterior intestine of the triatomine. These infective parasite forms released within the faeces of the triatomine can invade new vertebrate cells and undergo another round of differentiation into the intracellular amastigote forms, which proliferate and then transform back to trypomastigotes, the form that disseminates the infection. CD still represents a serious health problem in Latin America, with an overall prevalence of about 12-16 million cases [1]. In humans, its acute phase is mainly oligosymptomatic involving sometimes flu-like symptoms; this phase is associated with low mortality levels (<8%), especially among children. In the chronic phase, although most patients remain asymptomatic, after several months and even decades, about 30-40% of them may develop clinical symptoms characteristic of this phase, mainly cardiac and or digestive alterations [2].This infection is the leading cause of infectious cardiac disease in endemic areas [3]. The transmission of CD occurs primarily via the vector (90%), but may also occur through blood transfusion and congenital transmission, in addition to laboratory accidents [4], organ transplantation [5], and ingestion of contaminated food and beverages [6]. Recently, this disease has also been recognised as an opportunistic disease in HIV-infected individuals [7]. Furthermore, it is now being reported throughout the world due to growing international immigration [8].
Despite effective efforts focused on vectorial and blood transmission control, which have resulted in an important decrease in the number of new infected cases, many challenges still need to be faced: (i) its peculiar epidemiology, characterised by a variety of risk factors (many potential vectors and reservoirs, different forms of transmission and diverse parasite isolates present in domiciliary, peridomiciliary and sylvatic environments) and, importantly, (ii) the lack of prophylactic therapies and effective chemotherapeutic schemes [9].
Nifurtimox (Nf) and benznidazole (Bz) are the available drugs for the treatment of chagasic patients; however, due to their well-known toxicity and limited effect towards different parasite isolates and disease phases, new drugs are urgently needed [10-13]. Nf is currently produced and used predominantly in Central America and thus not available in Brazil. One of the main drawbacks of this drug is the high incidence of side effects, observed in up to 40% of patients, which include nausea, vomiting, abdominal pain, weight loss and severe anorexia in addition to adverse neurological effects, such as restlessness, paresthesias, twitching, insomnia and seizures [14]. Compared to Nf, Bz has the advantage of lower side-effect rates, which include hypersensitivity (dermatitis, generalised oedema, ganglionic infarction and joint and muscle pains), bone marrow depletion and peripheral polyneuropathy [15]. Bz production by LAFEPE (Laboratório Farmacêutico do Estado de Pernambuco, Recife, Brazil) will also include a new paediatric formulation recently supported by DNDi (Drugs for Neglected Diseases initiative) efforts. Regarding efficacy, both Bz and Nf provide parasite eradication in the acute phase of infection, although they require long-term therapies (up to 60 days), but neither are active in the prevalent chronic stage of the disease [10,16]. From the knowledge regarding the efficacy vs. toxicity of both nitro-heterocyclic compounds, the current recommendations for their use in CD include the treatment of all acute cases (including transfusion and laboratory-acquired disease and reactivation of infection in transplanted and other immunosuppressed patients) and recent chronic cases (including children up to 12 years of age) [14]. On the other hand, the contra-indications of Bz and Nf treatment include pregnancy, liver and kidney failure, neurological diseases unrelated to CD, and advanced CD with grade III or IV cardiopathy (OPS/OMS), as well as other pathologies that may be worsened by the treatment [15].
In fact, despite their undesirable side effects and the existence of parasite strains naturally resistant to both compounds, the major limitation of Bz and Nf is their insufficient anti-parasitic activity in the chronic form of the disease because approximately ≥ 80% of the treated chronic patients are not parasitologically cured [12,16]. On the other hand, clinical evidence suggests that, although not cured, the protective role of Bz treatment of chronic chagasic patients may achieve a significant reduction in the occurrence of electrocardiogram (ECG) changes and a lower frequency of deterioration of their clinical condition [17-24]. However, there is no consensus regarding the therapy of these chronic patients. As reported, the decision is left to the individual physician, as there is no definitive evidence arguing for or against such therapy, especially for those that already exhibit symptoms or signs of cardiopathy. In this sense, a project called Benznidazole Evaluation for Interrupting Trypanosomiasis (BENEFIT) is currently in progress and represents an international, multicentre, double-blind, placebo-controlled trial performed in Argentina, Bolivia, Brazil and Colombia with about 1,600 chronic chagasic patients with heart disease. BENEFIT aims to evaluate whether Bz treatment is able to significantly reduce parasite burden and addresses the unanswered question of whether etiologic treatment is beneficial for patients already presenting clinical manifestations of Chagas heart disease. In addition, BENEFIT aims to further explore the safety and tolerability profile of Bz [14].
Despite the fact that since 1912, many attempts have been made to treat the disease and some promising targets have been reported, from the time that Nf and Bz was introduced (1960-1970), only allopurinol and a few antifungal agents, including ergosterol biosynthesis inhibitors such as itraconazole, fluconazole and ketoconazole, have moved to clinical trials [25,26]. This situation may reflect (a) low investments by pharmaceutical companies, (b) mistaken concepts regarding the absence of parasitism during the chronic phase, and (c) the lack of standard procedures for drug screening. At present, a desired drug candidate for CD would include the following: (i) high activity against the parasite forms present in the mammalian hosts and against different T. cruzi stocks, (ii) efficacy against both acute and chronic infections, (iii) oral administration in few doses, (iv) low toxicity and improved safety (including children and women of reproductive age), (v) low cost and high stability for a long shelf life in tropical temperatures, and (vi) high levels of tissue accumulation and long term half lives [27].
Thus, as alternative drugs are desperately needed to treat CD, this article presents an overview of the biological activity, toxicity, selectivity and cellular targets of three classes of trypanocidal compounds - diamidines and related compounds, natural naphthoquinone derivatives, and megazol derivatives - that have been studied at the Laboratório de Biologia Celular (Instituto Oswaldo Cruz, Fiocruz, Brazil).
2. DIAMIDINES AND RELATED COMPOUNDS
Aromatic diamidines are DNA minor groove binders that have long been used in infectious disease chemotherapy [28,29]. Their major drawback is poor oral bioavailability and unfavourable side effects; thus, aiming to overcome these problems, new dicationic analogues and prodrugs have been synthesised from different medicinal chemistry groups and largely assayed in vitro as well as in vivo [30]. One of these examples is the prodrug of furamidine (DB75), named DB289, that has been in phase III clinical trials against human African trypanosomiasis. Unfortunately, despite initial indications of low toxicity in African, Asian, Caucasian and Hispanic populations, recent studies using extended dosages led to a withdrawal of DB289 from further human trials [13].
In recent years, our laboratory has been evaluating the biological effect of aromatic diamidines and analogues upon T. cruzi. We found that although DB75 and its N-phenyl substituted analogue DB569 (Fig. 1) display equivalent DNA-binding properties, the latter exhibited higher in vitro activity in the low micromolar range against different strains and stages of the parasite [31]. In T. cruzi-infected mice, DB569 led to reduction of the parasite cardiac load, down-modulation of cardiac CD8+ T cell levels, and reversion of the ECG alterations induced by the experimental acute infection [32]. The ECG protection provided by DB569 was also found during the chronic infection. The reversion to and further maintenance of a normal ECG profile in the DB569-treated mice may be associated with the reduced cardiac CD8+ lymphocyte infiltration and decreased parasitism, which might be ultimately contributing to higher mouse survival rates as compared to infected and untreated animals [33]. Additionally, as a higher trypanocidal effect of DB569 was also found during the incubation of T. cruzi-infected macrophage cultures as compared to the same treatment of cardiac cell cultures [31], it is possible that diamidines may also be acting directly on the immune cells. This subject is highly exciting because, besides controlling the parasite proliferation, these dicationic compounds may also regulate the inflammatory response and thus modulate the extent of tissue damage induced by an unbalanced immune response.
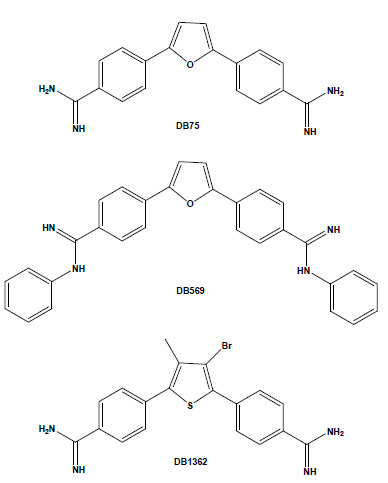
Chemical structures of the aromatic diamidines DB75, DB569 and DB1362.
Another study performed using both amastigote and trypomastigote forms showed the in vitro efficacy of the diarylthiophene diamidine DB1362 (Fig. 1) at micromolar doses that did not cause any toxicity to the host cell [34]. This diamidine showed good trypanocidal effect at 4°C in the presence of plasma constituents (mouse blood), exhibiting similar IC50 values as those found at 37°C in the absence of blood, arguing for its possible application in prophylaxis of blood banks. In vivo assays performed by the administration of only 2 doses of DB1362 at 25 mg/kg (the first at the parasitaemia onset and the second at the peak) reduced the parasitaemia levels by 40% and resulted in 100% protection against mortality induced by the acute infection in this experimental model. As previously observed with DB569, DB1362 also largely reduced cardiac parasitism (similar levels as Bz run in parallel) in addition to protecting against electric cardiac alterations due to T. cruzi infection [34].
Recently, the biological and ultrastructural effects and subcellular localisation of another series of synthetic heterocyclic dicationic compounds were studied in T. cruzi [35]. These diamidines were selected aiming to determine their efficacy on both bloodstream trypomastigotes and intracellular forms of the parasite. These compounds can be divided into three main groups: (i) curved molecules the size of DB75 or larger (DB1645, DB1651), (ii) a curved molecule smaller than DB75 (DB1582) and (iii) linear molecules smaller than DB75 (DB1627, DB1646, DB1670). The data showed that all compounds exerted low toxicity to mammalian cells (LC50 > 96 µM). However, although the first two groups (DB1645, DB1651 and DB1582) showed good trypanocidal activity, providing IC50 values in the range of 0.15 to 13.3 µM, the smaller linear molecules were not effective [35].
Arylimidamides (AIAs), also previously known as reversed amidines, have extraordinary activity against both trypanosomatids, such as T. cruzi [36,37], and Leishmania spp. [38-40]. In contrast to the original diamidines, in which the imino group is directly attached to an aryl ring, AIAs present the imino group attached to an “anilino” nitrogen [39]. AIAs, such as DB889, present very high activity (at sub-micromolar and nanomolar levels) against intracellular amastigotes and bloodstream forms of different stocks of the parasite and also show superior efficacy to diamidines as well as other closely related compounds [41]. It is possible that the higher efficacy of AIAs as compared to diamidines may be related to their physical properties because while amidines are highly basic molecules with pKa values near 11, the pKa values of AIAs are near 7. Thus, at physiological pH, amidines are protonated, cationic molecules, whereas the arylimidamides are essentially neutral. This difference may significantly affect not only absorption, but also distribution, and likely plays an important role in the differences in activity between AIAs and diamidines [35]. In another recent study, we reported the in vitro trypanocidal effects of several different heterocyclic cationic compounds, including diamidines, a monoamidine, an arylimidamide and a guanylhydrazone, and found that all compounds exerted, at low micromolar doses, a time-dependent trypanocidal effect to varying extents upon both intracellular parasites and bloodstream trypomastigotes [37]. Among them, the higher potency and selectivity of the AIA DB613A (Fig. 2), especially towards intracellular parasites (reaching a selective index of >126), corroborate the highly promising activity of AIAs against this parasite, which further justifies studies in experimental models of T. cruzi infection. Additionally, AIAs such as DB702 and DB889 (Fig. 2) also present very high trypanocidal activity against bloodstream forms incubated at 4°C in the presence of blood sources (100% mouse blood), also showing the possible application of AIAs in the prophylaxis of blood banks [41].
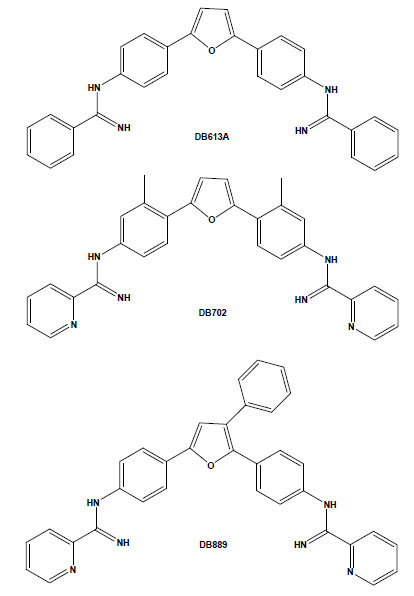
Chemical structures of the arylimidamides DB889, DB702 and DB613A.
Several potential transporters that effectively carry diamidines have been reported in some parasites, including African trypanosomes, Leishmania spp. and Plasmodium falciparum [42-44]. However, their uptake mechanisms in T. cruzi are still unknown and deserve investigation. On the other hand, the intrinsic fluorescence of most of these dicationic aromatic compounds allows following their uptake and sub-cellular accumulation within the treated cells. Aromatic diamidines were localised in DNA-enriched organelles, such as nuclei, and, more importantly, within the mitochondrial kDNA of T. cruzi [31,35,45]. In addition to the usual localisation of these compounds in the parasite nuclei and kDNA (with greater intensity in the latter structure), some diamidines, such as DB1582 and DB1651 (Fig. 3), were also found within punctate non-DNA-containing cytoplasmic organelles, possibly acidocalcisomes, although other non-DNA-containing organelles cannot be excluded [35]. It is possible that, as suggested for the African trypanosomes [46], the localisation of these compounds within these acidic organelles could play a role in their mechanism of action and/or act as storage sites in T. cruzi, and additional studies are necessary to clear up this matter. An important point to be considered is the unusual disorganisation of microtubules, with the occurrence of multiple axoneme structures observed in diamidine- and AIA-treated bloodstream trypomastigotes [35,41]. Up to now, no major alterations have been reported in both sub-pellicular and flagellar microtubules of T. cruzi after incubation with different classes of drugs that target microtubules, such as taxol, colchicine and vinblastine, possibly due to the high content of acetylated tubulin and/or poly-glutamylation of tubulin found in these parasites [47,48]. Because these structures in trypanosomatids are highly resistant to microtubule disrupters as compared to those in mammalian cells, they represent interesting targets for drug development and deserve further investigations regarding the alterations induced by these dicationic aromatic compounds.
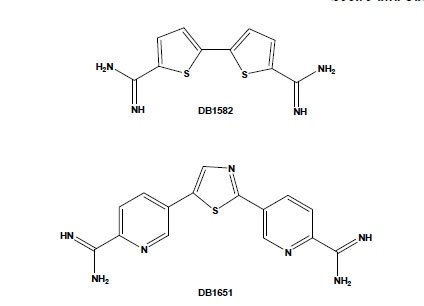
Chemical structures of the aromatic diamidines DB1582 and DB1651.
In fact, although the exact mechanism of action of these heterocyclic dications upon T. cruzi as well as upon other trypanosomatids has not been clearly demonstrated, it is likely that multiple modes of action may be operative and that compound uptake represents a fundamental step in their action and contributing, in part, to their selectivity (Wilson et al. 2008; de Koning, 2001). On the other hand, several data suggest that at least part of the activity is related to their selective binding to AT-rich sequences of kDNA minicircles, leading to the destruction of the kinetoplast and parasite death [29,49]. In fact, transmission electron microscopy studies showed that the structure of parasite mitochondria (and kinetoplasts) is highly altered by aromatic diamidines and by some AIAs at doses that do not affect mammalian host cells [31,35,41,45]. Flow cytometry studies confirmed that diamidines and AIAs target the mitochondria-kinetoplast complex of T. cruzi through the interference of these compounds with the proton electrochemical potential gradient of the mitochondrial membrane of the parasite [34,41] and that diamidines, such as DB569 and DB75, trigger apoptosis-like death in bloodstream forms of T. cruzi [32]. However, the in vitro screening of 11 dicationic aromatic molecules against T. cruzi showed that, in agreement with previous data reported in T. brucei [50], although all were localised to a higher extent within the kinetoplast than in the nucleus, no correlation could be found between compound activity and higher kDNA accumulation [45]. Additionally, it has been proposed that although diamidines are excellent minor groove DNA binders, this interaction itself could not fully explain their biological activity. Recent reports suggest that their association with DNA could represent an initial step, followed by topological changes leading to molecule instability and destruction and/or modification of DNA-protein interactions, leading to replication errors, DNA degradation and parasite death [29,51]. Other targets that have also been proposed include inhibition of tyrosyl-DNA phosphodiesterase, topoisomerases, protein kinase A, proteases and polymerases [52-54].
In summary, the efficacy of these DNA binders, especially AIAs, against T. cruzi stimulates further studies aiming to help establish a valuable scheme of therapy for CD.
3. NATURAL NAPHTHOQUINONE DERIVATIVES
Naphthoquinones are compounds present in different families of plants. Their molecular structures endow them with redox properties, and they are involved in different biological oxidative processes. The biological redox cycle of quinones can be initiated by one electron reduction leading to the formation of semiquinones, unstable intermediates that react rapidly with molecular oxygen, generating free radicals. Another alternative mechanism is reduction by two electrons, mediated by DT-diaphorase, leading to the formation of hydroquinone. These compounds act as inhibitors of electron transport, uncouplers of oxidative phosphorylation, DNA intercalating agents, and producers of reactive oxygen radicals [55-57], being considered privileged structures in medicinal chemistry studies.
Lapachol is easily isolated from the heartwood of trees of the Bignoniaceae family, while both α-lapachone and β-lapachone are present only in small amounts. Previous reports showed that against T. cruzi epimastigotes, β-lapachone increases the generation of reactive oxygen species through formation of the semiquinone radical, leading to lipid peroxidation and inhibition of nucleic acid and protein synthesis [58-62]. T. cruzi is known to be deficient in reactive oxygen and nitrogen species detoxification, being especially sensitive to oxidative stress conditions [63], and its single mitochondrion, containing a branched electron transport chain and a specialised region rich in DNA named kinetoplast [52], is an extraordinary drug target [27]. The ultrastructural injuries observed in β-lapachone-treated epimastigotes [64] together with the increase in the generation of hydrogen peroxide clearly demonstrates the mitochondrial susceptibility of T. cruzi to naphthoquinones.
These naphthoquinones were used as starting points for medicinal chemistry studies, and through their reaction with amino-containing reagents, 14 oxazoles, 30 imidazoles, and 10 other related heterocyclic compounds were synthesised. The reaction of 1,2-naphthoquinones with aromatic aldehydes led to naphthooxazoles and naphthoimidazoles, easily separated by column chromatography, and in several instances, the latter type of derivative was obtained as the main product and easily isolated by vacuum filtration and crystallisation in hexane/ethyl acetate. The comparison of their activity on bloodstream trypomastigotes revealed that structural features that produced an increase in lipophilicity, such as a furan moiety, methoxyl group, or an aliphatic side chain, may have contributed to the increase in the anti-T. cruzi effect [65]. Among the 80 compounds screened against trypomastigote forms, the most active were three naphthoimidazoles derived from β-lapachone with the aromatic moieties phenyl (N1), 3-indolyl (N2) and p-methylphenyl (N3) (Fig. 4) [66-69], pointing to the tendency of trypanocidal activity in compounds in which aromatic structures were introduced at the imidazole ring linked to a naphthopyranic structure, a promising route in the search of heterocycles with potential activity against T. cruzi. The naphthoimidazoles N1, N2 and N3 were also active against intracellular amastigotes and epimastigotes of T. cruzi and presented low toxicity to host cells [70]. In epimastigotes, the compounds blocked the cell cycle and metacyclogenesis, inhibited succinate cytochrome c reductase and induced damage to the mitochondrion, Golgi complex and reservosomes. In trypomastigotes, the compounds caused alterations in the kinetoplast and mitochondrion in addition to inducing blebs in the plasma membrane and DNA fragmentation [71-73]. The mitochondrion, reservosomes and DNA were identified as the main targets in T. cruzi. The compounds’ precise mode of action upon the parasite appears to be complex; however, we can exclude damage caused by oxidative stress because, unlike the original naphthoquinones, N1, N2 and N3 do not easily undergo redox reactions. It is important to note that several trypanocidal agents, such as benznidazole, contain imidazole moieties [74-76], which is consistent with the idea that the trypanocidal activity is associated with the imidazole skeleton.
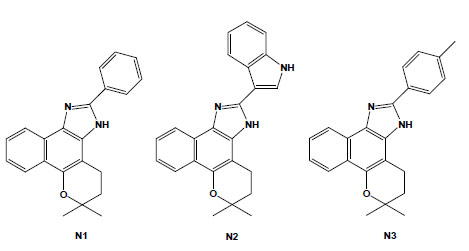
Chemical structures of naphthoimidazoles derived from β-lapachone. N1 [4,5-dihydro-6,6-dimethyl-6H-2-(phenyl)-pyran[b-4,3]naphth[1,2-d]imidazole]; N2 [4,5-dihydro-6,6-dimethyl-6H-2-(3′-indolyl)-pyran[b-4,3]naphth[1,2-d]imidazole]; and N3 [4,5-dihydro-6,6-dimethyl-6H-2-(4′-methylphenyl)-pyran[b-4,3]naphth[1,2-d]-imidazole].
Taking into account that molecular hybridization is a powerful approach for the design of new compounds based on the recognition of the pharmacophoric sub-unities in the molecular structure, new quinone derivatives were prepared exploring the electrophilicity of 1,2-quinoidal carbonyls by reactions with different heteroatom nucleophilic centres. In this context, different classes of derivatives were obtained by the hybridisation of naphthoquinones with [1,2,3]-triazoles or arylamines and screening as anti-T. cruzi compounds. Compounds based on [1,2,3]-triazole comprise an important class of aromatic heterocycles with a wide range of biological and pharmacological activities. Their reaction with nor-β-lapachone by Huisgen cyclisation under copper catalysis led to derivatives that were shown to be more active against T. cruzi than the parent quinone, with IC50/1 day values in the range of 17 to 359 mM, with the apolar phenyl-substituted triazole 6 being the most active (Fig. 5) [77]. From the reaction of nor-lapachol with aryl amines, substituted ortho-naphthofuranquinones were created, and two compounds, the 4-methoxy- (NF1) and the 3-nitrophenylamino (NF2) derivatives, were more active than benznidazole, the standard drug [78]. From C-allyl lawsone, three naphthofuranquinones (NF3, NF4 and NF5; Fig. 5) were also assayed, showing activity against trypomastigote, intracellular amastigote and epimastigote forms in addition to presenting low toxicity to the host mammalian cells [79]. Although there are several reports on the biological activity of naphthopyranquinones, data on naphthofuranic analogues are scarce. Ortho-naphthoquinones, such as β-lapachone, have already been reported to present higher trypanocidal activity in comparison to their para isomers [66]. NF3, which is a para derivative, was more active than the ortho-quinones NF4 and NF5. The three compounds were active against intracellular amastigotes, causing a strong decrease in the percent of infection, while damage to the host cells was observed only in concentrations 10-fold higher than the doses effective against the parasite. Ultrastructural analysis of treated epimastigotes and trypomastigotes indicated a potent effect of the compounds on the mitochondria, which were significantly swollen and possessed a washed-out matrix profile [80]. NF3, NF4 and NF5 also led to a collapse of the mitochondrial membrane potential and specifically decreased mitochondrial complex I-III activity in both epimastigotes and trypomastigotes, which paralleled the reduction in succinate-induced oxygen consumption. The interference with electron flow at the inner mitochondrial membrane by reduced naphthoquinones suggests that reactive oxygen species generation could promote a functional impairment of the mitochondrion because this organelle is crucial in the trypanocidal effect of these naphthofuranquinones in T. cruzi [80].
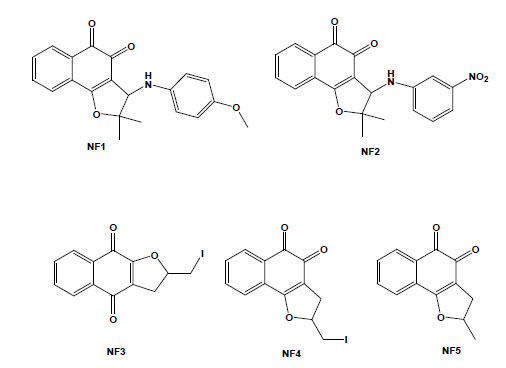
Chemical structures of natural naphthoquinone derivatives: NF1 [3-(4-methoxyphenylamino)-2,3-dihydro-2,2-dimethylnaphtho [1,2-b]furan-4,5-dione]; NF2 [3-(3-nitrophenylamino)-2,3-dihydro-2,2-dimethylnaphtho[1,2-b]furan-4,5-dione]; NF3 [2,3-dihydro-2-iodomethylene-4,9-dioxonaphtho[2,3-b]furan]; NF4 [2,3-dihydro-2-iodomethylene-4,5-dioxo-naphtho[1,2-b]furan]; and NF5 [2,3-dihydro-2-methyl-4,5-dioxo-naphtho[1,2-b]furan].
Such hybrid molecules endowed the quinones with redox properties, representing an interesting starting point for a medicinal chemistry program directed toward the chemotherapy of CD. Their facile synthesis opens the possibility of large-scale production with high yields that can be assayed in experimental mouse models.
4. MEGAZOL DERIVATIVES
The 5-nitroimidazole megazol was shown to be highly active in vitro and in vivo against T. cruzi, including strains resistant to benznidazole [81-84], and has become a core structure for the design of new leads for the treatment of CD. Megazol has been described as a trypanothione scavenger, the cofactor for trypanothione reductase [85,86]. Despite its noteworthy trypanocidal activity, megazol development was discontinued due to reports of mutagenic and genotoxic in vitro effects [87-89]. In the late 90s, with its resurgence as a new potential drug for African trypanosomes [90,91] and trying to circumvent its toxic profile, megazol began to be explored as a template for the design of new trypanocidal drugs [92-94].
From the in vitro screening using trypomastigote forms, eight 1,3,4-thiadiazole-2-arylhydrazones out of thirty-four synthesised megazol derivatives were selected based on their potent trypanocidal activities [95,96]. These compounds were prepared exploring the hypothesis that the introduction of a radical scavenger arylhydrazone moiety in the heterocyclic framework of megazol could modulate the production of toxic nitro anion radical species, avoiding potentially mutagenic properties [97,98]. Guanylhydrazones have been previously shown to display strong activity against T. cruzi [99]. It is also well known that some functionalised hydrazone-related derivatives [100-102] presented trypanocidal activity, which could be correlated with an action on essential enzymes of T. cruzi, such as cruzipain and trypanothione reductase, potential targets of new drugs for CD [27]. Such a strategy led us to identify a new potent trypanocide prototype S1 (Brazilizone A) (Fig. 6), which was two times more potent than megazol against the infective bloodstream form of the parasite and was selected for subsequent in vitro and in vivo studies. An additional 7 megazol derivatives also displayed interesting bioprofiles, with IC50s/24 h in the range of 11 to 63 µM: the 4-OH (S2); the lipophilic para-bromo (4-Br) (S3); the 4-NO2 (S4); 3,5-dit-Bu,4-OH (S5); the 4-Cl (S6); the 4-OMe (S7); and the 2-OH (S8) (Fig. 6) [103]. S1, S2 and S3 led to several ultrastructural alterations in trypomastigotes and were also active against intracellular amastigotes at concentrations that induced no toxicity to the host cell. T. cruzi-infected mice were treated with each of these three derivatives, and while S1 did not interfere with the course of infection, S2 led to a significant decrease of parasitaemia and S3 decreased mortality. In this context, as observed in the literature [104], there is not always a direct correlation between the in vitro effect on the clinically relevant trypomastigote and amastigote forms of T. cruzi and the results of the treatment in experimental models.
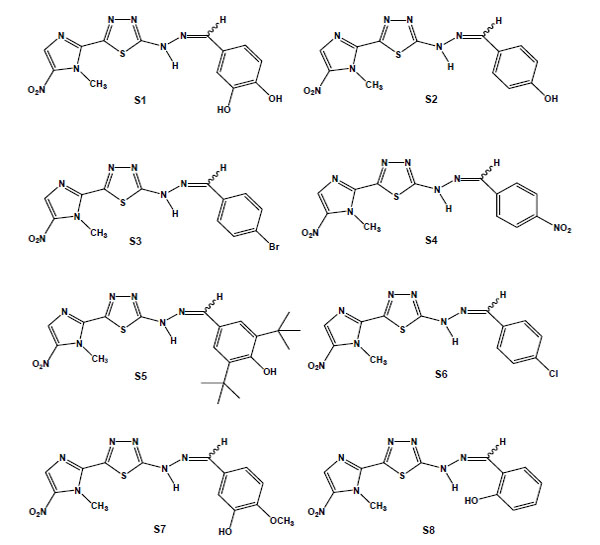
Chemical structures of megazol derivatives: S1, 3,4-dihydroxybenzylidene [5-(1-methyl-5-nitro-1H-imidazol-2-yl)-1,3,4-thiadiazol-2-yl]hydrazine); S2 (4-hydroxybenzylidene [5-(1-methyl-5-nitro-1H-imidazol-2-yl)-1,3,4-thiadiazol-2-yl]hydrazine); S3 (4-bromobenzylidene [5-(1-methyl-5-nitro-1H-imidazol-2-yl)-1,3,4-thiadiazol-2-yl]hydrazine); S4 (4-nitrobenzylidene [5-(1-methyl-5-nitro-1H-imidazol-2-yl)-1,3,4-thiadiazol-2-yl]hydrazine); S5 (3,5-ditertbutyl-4-hydroxybenzylidene [5-(1-methyl-5-nitro-1H-imidazol-2-yl)-1,3,4-thiadiazol-2-yl]hydrazine); S6 (4-chlorobenzylidene [5-(1-methyl-5-nitro-1H-imidazol-2-yl)-1,3,4-thiadiazol-2-yl]hydrazine); S7 (3-hydroxy-4-methoxy-benzylidene [5-(1-methyl-5-nitro-1H-imidazol-2-yl)-1,3,4-thiadiazol-2-yl]hydrazine); and S8 (2-hydroxybenzylidene [5-(1-methyl-5-nitro-1H-imidazol-2-yl)-1,3,4-thiadiazol-2-yl]hydrazine).
5. FINAL COMMENTS
For more than 15 years, our group has been investigating the biological activity, toxicity, selectivity and cellular targets of different classes of compounds upon T. cruzi, in collaboration with research groups focused in medicinal chemistry. An overview of in vitro and in vivo studies dealing with aromatic diamidines, naphthoquinones and megazol derivatives was presently discussed, aiming to contribute for the current knowledge and development of new arsenal candidates with anti-T. cruzi efficacy. In our investigation of the mechanism of action of trypanocidal compounds an important tool is electron microscopy for the determination of cellular targets in the parasite. We observed that aromatic diamidines and naphthoquinone derivatives interact with T. cruzi DNA, promoting a remarkable chromatin condensation and fragmentation [70,71] and in some cases, leading to nuclear envelope dilation [80], at doses that do not affect mammalian host cell viability. Another DNA-enriched compartment is the unique mitochondrion of trypanosomatids which is deficient in reactive oxygen species detoxification, and as so an attractive drug target. Treatment with aromatic diamidines and naphthoquinone derivatives induced a severe mitochondrial swelling with loss of functionality confirmed by flow cytometry, fluorescence microscopy and biochemical approaches [31,34-36,70,71,80]. It is important to keep in mind, however, that induced mitochondrial alterations may be due either to a primary effect directly on this organelle or secondary lesions caused by loss of cellular viability triggered by another cell component or metabolic pathway. Aromatic diamidines and megazol derivatives were shown to be active in vitro and in vivo against T. cruzi [32-34,103], however, interestingly, there is not always a direct correlation between the in vitro effect on trypomastigote and amastigote forms and that of the treatment in experimental models.
In the last decades only few compounds moved to clinical trials for CD possibly due to the low rates of investments alocated in this area and to the lack of standardized protocols for drug screening. The peculiar epidemiologic characteristics of CD (including different hosts, geographic environments, transmission routes and cycles, developmental forms and strains) require a continuous and appropriate evaluation on the disease control tools and keep this neglected disease as a relevant challenge for the health ministry of many developing countries on the American continent. The use of etiologic treatment not only in the acute phase, but also in chronic patients is strengthened by the growing evidence that the low-grade, incessant myocarditis seen in the later stage of CD is related to parasite persistence, along with unbalanced immune responses in some individuals, leading to a sustained inflammatory response that underlies the characteristic lesions of chronic CD. Thus, the current concept is that parasite elimination and/or its burden decrease may hinder the outcome of this parasitic disease [12,14]. In this context, as no safe and fully effective drug is available, a collaborative interdisciplinary network for pre-clinical studies on drug discovery is combining parasitology and cellular biology expertise, medicinal chemistry, SAR patterns, physical chemical properties analysis and pharmacological studies associated with standard procedures and harmonised experimental in vitro and in vivo models. Promising targets for Chagas chemotherapy includes kDNA, sterol metabolism, and parasite specific enzymes such as cysteine proteinase [27]. The knowledge of the crystal structure and the specificity of the major T. cruzi cysteine protease, cruzipain, has allowed the design of specific peptidyl as well as non peptidyl inhibitors [10,11]. Among the extensive list of compounds that have been screened against T. cruzi in the last three decades, ergosterol biosynthesis inhibitors represent the most advanced candidates for new anti-T. cruzi agents, and among them, some new triazole derivatives that are able to inhibit the parasite’s C14α sterol demethylase are the most promising. Two of them, posaconazole (Schering-Plough Research Institute) and ravuconazole (Eisai Company), are poised for clinical trials in the short term, although the cost of manufacture of these new compounds may represent an obstacle [12]. However, until an ideal novel drug for the specific treatment of CD is identified, the use of alternative therapies, such as the use of combined therapy using Bz and Nf associated with old drugs already available in the market for other pathologies, represent a promising approach [15]. In addition, it is relevant to state the necessity to provide an all-around care through adoption of a biopsychosocial model considering the patient therapy in the context of the biological, psychological and social factors [105]. As previously pointed out, “…the more leads/approaches that progress to investigational drug candidates, the better chance that new treatments for this often fatal infection will be available to patients in the near future” [11].
ACKNOWLEDGEMENTS
Sponsored by CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico), Faperj (Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro), Fundação Oswaldo Cruz, CPDD (Consortium for Parasitic Drug Development) and CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior).

