All published articles of this journal are available on ScienceDirect.

Tricyclic Pyrazoles. Part 5. Novel 1,4-Dihydroindeno[1,2-c]pyrazole CB2 Ligands Using Molecular Hybridization Based on Scaffold Hopping
Authors Info & Affiliations
Abstract
In search of new selective CB2 ligands, the synthesis and preliminary biological evaluation of novel 1,4-dihydroindeno[1,2-c]pyrazole hybrids of the highly potent prototypicals 5-(4-chloro-3-methylphenyl)-1-(4-methylbenzyl)-N-fenchyl-1H-pyrazole-3-carboxamide 1 and 1-(2,4-dichlorophenyl)-6-methyl-N-(piperidin-1-yl)-1,4-dihydroindeno[1,2-c]pyrazole-3-carboxamide 2 are detailed.
We postulated that the introduction of those pharmacophoric elements essential for activity of 1 in the tricyclic core of 2 might provide CB2 ligands with further improved receptor selectivity and biological activity. Among the compounds, 6-chloro-7-methyl-1-(2,4-dichlorophenyl)-N-fenchyl-1,4-dihydroindeno[1,2-c]pyrazole-3-carboxamide (22) exhibited low two digit nanomolar affinity for the cannabinoid CB2R and maintained a high level of CB2-selectivity.
INTRODUCTION
Endocannabinoid ligands (such as anandamide [1] - AEA and 2-arachidonoylglycerol - 2AG, Fig. 1) [2, 3], which belong to the fatty-acid-derived neuromodulators, mediate their pathophysiological functions through at least two known G protein-coupled receptor subtypes, CB1 and CB2 receptors [4, 5] (CB1R and CB2R). Evidence for a third type of G protein-coupled cannabinoid receptor, CB3, in brain and in endothelial tissues is mounting [6-8]. However, the cloning, expression and characterization of CB3 has yet to be reported.
Structures of Anandamide and 2-Arachidonoylglycerol.
The CB2R has been shown to be present in peripheral tissues (tonsils, thymus, spleen, pancreas), peripheral nerve terminals, skin tumor cells [9-11] and on cells of the hematopoietic lineage such as inflammatory cells, eosinophils, monocytes, basophiles, natural killer cells and T and B cells [12-17]. Furthermore, there is a growing body of evidence that is based on animal models in which the CB2R plays a role in immune and inflammatory responses [16-20]. However, CB2 gene transcripts and receptors have also been discovered in the central nervous system (CNS). The expression of CB2Rs in the brain suggests that they may play broader roles in the CNS than previously appreciated [21].
Because of the therapeutic potential of CB2 ligands, especially for the treatment of cancer [22, 23], multiple sclerosis [24-26], Alzheimer’s disease [27], neuropathic and inflammatory pain [28-31], and diabetes [32], there has been an increasing interest in the development of potent ligands of this receptor. To date several structural classes of cannabinoid ligands have displayed affinity and selectivity for the CB2R [33-43]. In these preceding studies the relevance and structure-activity relationships (SAR) of pyrazole derivatives has been consolidated and clarified, and in the process compounds 1 (SR144528) [44] and 2 [45] emerged as important prototypical CB2 selective ligands for further studies (Fig. 2). It is plausible to think that the interaction points shown in Fig. (2), and particularly the fenchyl group at the C3 position of compound 1, may be involved in preferential binding at the CB2R, accounting for its in vitro activity as both a potent and selective (> 600-fold) CB2 ligand and its remarkable in vivo CB2R antagonist activity in CB2R-mediated gastrointestinal assays [44]. An additional breakthrough in identifying CB2 selective ligands was the discovery that the flattening of the plane of the tricyclic core of the compound 2 was a determining factor in its very high CB2-affinity (Ki = 0.037 nM), exceptional selectivity over CB1R, and CB2 agonist activity in vitro [45].
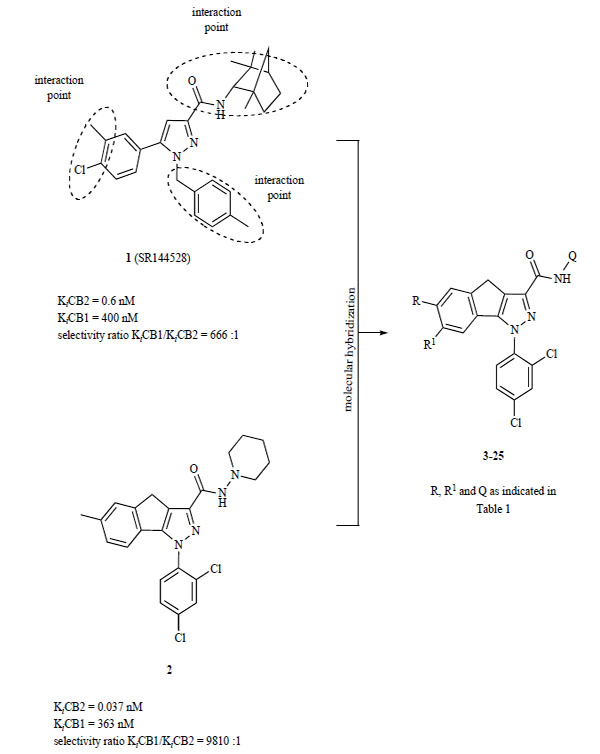
Structural design concept of compounds 3-25.
A limited SAR study on congeners of 2 has recently been explored but none of these analogues displayed higher CB2-affinity respect to the lead [45]. The lack of available crystal structure of the cannabinoid receptors necessitated ligand-based design, in which compounds possessing pharmacophoric elements consistent with cannabinergic activity serve as the basis for creation of new ligands [46].
Based upon the putative interacting sites and structural features of 1 (i.e., the N1-aryl group, the C3 carboxamide moiety, and the substitution of the C5 phenyl ring), we postulated that the introduction of those pharmacophoric elements in the tricyclic core of 2 might provide CB2 ligands with further improved receptor selectivity and biological activity. Thus novel compounds that varied two of these components, the phenyl substitution and the carboxamide moiety, were designed (Table 1). We report syntheses of these hybrid compounds 3-25 together with preliminary aspects of their receptor affinity, selectivity and biological activities.
Structure and Binding Data for Compounds 3-25
 |
CHEMISTRY
The new CB2 ligands 3-25 were synthesized starting from ketones 26 [45] and 27 [47] (Scheme 1). Their α-acylation by Claisen reaction furnished the α-keto-γ-hydroxyesters 28, 29 in good yields. The cyclization with 2,4-dichlorophenyl hydrazine hydrochloride in refluxing AcOH gave the tricyclic dihydroindeno[1,2-c]pyrazole esters 30, 31, which hydrolysis with potassium hydroxide in hydroalcoholic solution afforded acids 32,33 in quantitative yields. Target compounds 3-17,19-25 were synthesized by condensation of acids 32, 33, previously activated with 1.1 equivalents of 1-hydroxybenzotriazole (HOBt)/1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC), in dichloromethane with the appropriate amines, where as only for compound 18 was necessary the formation of the acyl chloride of acid 32, then the condensation with 1-naphthylamine.

Reagent and conditions: a) Na, dry EtOH, (COOEt)2, RT, 24 h; b) 2,4-Cl2-C6H3NHNH2·HCl, AcOH, rfx, 8 h; c) KOH, EtOH/H2O, rfx, 4 h; d) HOBt/EDC, CH2Cl2, RT, 1 h, then Q-NH2, (TEA for 11 and 20), CH2Cl2, RT, 22 h; (e) SOCl2, toluene, rfx, 3 h (for 18), then 1-naphthylamine, CH2Cl2, RT, overnight.
RECEPTORIAL AFFINITY AND SELECTIVITY
To evaluate the affinity of the synthesized compounds towards CB1R and CB2R, radioligand binding assays were carried out by competition with [3H]-CP-55,940 in mouse brain (minus cerebellum) and spleen homogenates, respectively. The assays were performed according to previously reported procedures [45].
Intrinsic activity of the new compounds characterized by the highest affinity towards CB2 receptors was preliminary determined through an in vitro model based on phosphorylated ERK ½ (P-ERK ½) expression detection by treating human promyelocytic leukemia HL-60 cells [45]. In fact it has been reported that this cell line selectively expressing CB2R but not CB1R. Moreover P-ERK ½ activation is induced on HL-60 cells by CB2 agonist derivatives and the effect of these compounds is counteracted by the CB2 selective antagonists SR144528 or AM630.
RESULTS AND DISCUSSION
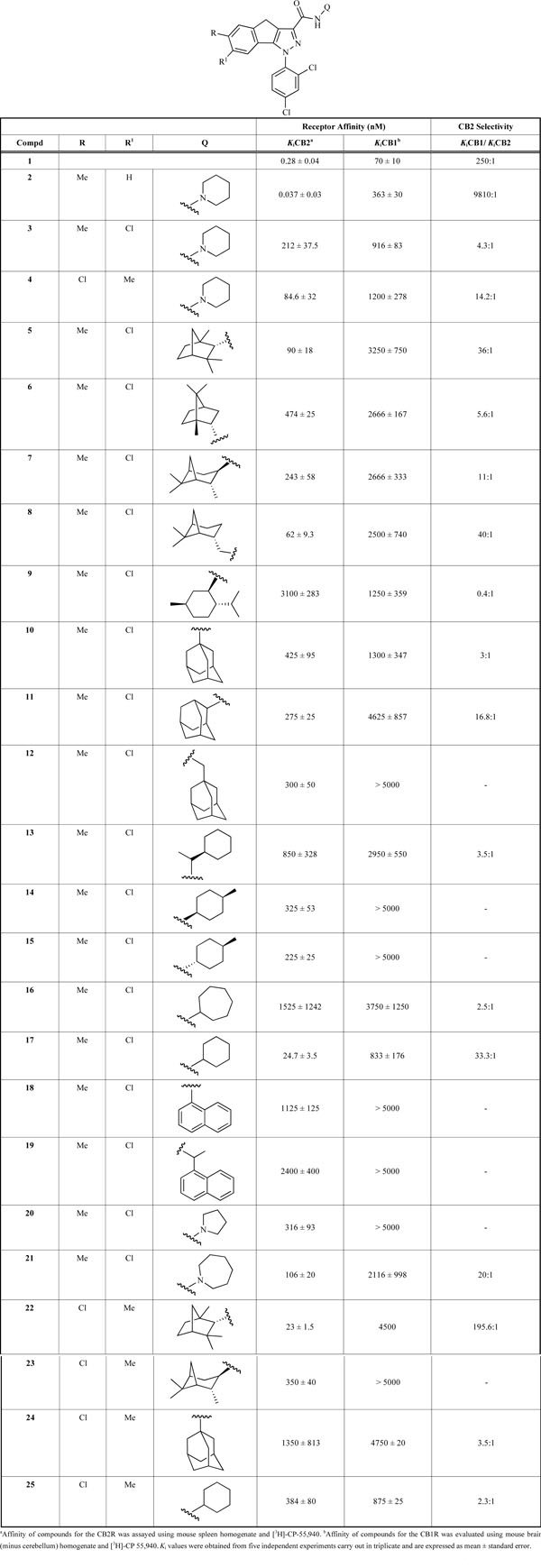
The cannabinoid receptor affinities of the new 1,4-dihydroindeno[1,2-c]pyrazoles 3-25 are shown in Table 1. For comparison, the Ki values of the lead compound 6-methyl-1-(2,4-dichlorophenyl)-N-piperidin-1-yl-1,4-dihydroindeno [1,2-c]pyrazole-3-carboxamide (2) and reference CB2 ligand SR144528 (1) have been reported. Results are the average of five independent experiments with three replicates at each concentration.
The initial introduction of the chlorine group at the R1 position of lead 2 gave compound 3 [45] which showed a decrease in CBRs affinity (KiCB2 = 212 nM; KiCB1 = 916 nM) and CB2 selectivity (KiCB1/KiCB2 = 4.3) as compared to compound 2. To further estimate the influence on the CBRs-affinity of the addition of the chlorine group in the tricyclic scaffold of 2, we synthesized compound 4 [45] in which the substituents R and R1 of 3 were exchanged: this change produced a similar decrease in CBRs-affinity (KiCB2 = 84.6 nM; KiCB1 = 1200 nM) and subtype receptor selectivity (KiCB1/KiCB2 = 14) as previously observed for 3.
In light of the results obtained with these modifications in the phenyl ring of the tricyclic system in compounds 3 and 4, it was of interest to further determine the influence of the addition of a fenchyl group at the C3 carboxamide portion, mimicking compound 1, with the aim to evaluate the effects on CB2-affinity and selectivity of such modification on the dihydroindenopyrazole core. The replacement of the carboxamide N-piperidinyl moiety of 3 with the fenchyl residue gave the analogue 5 with enhanced CB2-affinity and improved subtype receptor selectivity (KiCB2 = 90 nM; KiCB1/KiCB2 = 36) compared to those of 3. Because the hybrid analogue 5 displayed reasonable CB2-affinity and selectivity, we decided to further explore the SAR of this ligand through the introduction of other bulky groups in the carboxamide moiety, which should provide an improved understanding of the structural features that influence the affinity of this novel tricyclic scaffold.
We first synthesized a small library of four monoterpene compounds (6, 7, 8 and 9) and three adamantine derivatives (10, 11 and 12). All these compounds, with the exception of 8, containing monoterpene and adamantaneamine-side moieties had, in general, reduced CB2-affinity compared to that of compound 5 (KiCB2 about 3-5 times higher). However, excluding 9 which posses KiCB2 value of 3100 nM, all the compounds belonging to this subclass showed CB2-affinity equivalent to that of 3 (KiCB2 comprises between 243 and 474 nM). For example, compounds 8 (KiCB2 = 62 nM and KiCB1/KiCB2 = 40), containing an amine cis-myrtanyl substituent, had approximately 1.5 fold increased affinity as compared to 5. These results are of interest because they show that, among these derivatives, only this cis-myrtanyl derivative, improves the affinity for the CB2R. The monoterpene and adamantine derivatives, on the other hand, had generally very low affinity against CB1R.
To further explore whether improvements in CBR-affinity might be obtained by modifying the C3-carboxamide side group, we next synthesized the five analogues of 5, shown in Table 1, in which we simplified the skeleton of the amine side chain at the C3 carboxamide unit (compounds 13-17). Among these carbocyclic compounds, the cyclohexyl derivative 17, similar to our previously reported data concerning another series of tricyclic derivatives [45], displayed the highest affinity to CB2R (Ki = 25 nM). However the CB1/CB2 selectivity of 17 was again decreased to 4.7-fold lower than that determined for 2. It was also determined that replacement of the N-fenchyl moiety of 5 by the electron rich 1-naphthyl (18) and the 1-naphthylmethyl tail-pieces (19) resulted in compounds with very low affinity for both CB2R and CB1R.
In order to investigate the effect of the substitution of a relatively simple non aromatic heterocyclic ring containing a nitrogen atom at this position on the CBR-affinities, compounds 20 and 21 with N-1-pyrrolidinyl and N-1-homopiperidinyl substituents were examined. Intermediate Ki values for CB2-affinity were determined for the pyrrolidinyl (20, Ki = 316 nM) and homopiperidinyl (21, Ki = 109 nM) containing group derivatives. The N-homopiperidinyl substituted benzocyclopentapyrazole displayed some CB1/CB2 selectivity (KiCB1/KiCB2 = 20).
Finally, the influence of the R and R’ substitution pattern at the C3 position and the nature of the carboxamide moiety of 4 was examined (22-25). Compound 22, the direct analogue of 5 with R and R’ exchanged exhibited 4-fold more affinity for CB2Rs as compared to 4 and 5, while the affinity for the CB1R was slightly diminished. As a result, the 6-chloro-7-methyl-disubstituted analogue 22 had the highest CB2 to CB1 selectivity (KiCB1/KiCB2 = 195.6) among all tested compounds. Replacement of the fenchyl group of 22 with an isopinocampheyl (23), adamantyl (24) and cyclohexyl (25) group resulted in 16.7 - 58.6-fold loss in CB2R-affinity and no remarkable effect on CB1 binding affinity.
To summarize the results between the compounds bearing the same substituents at R and R’, it is plain that the introduction of a fenchyl group lead to an improvement of the CB2R-affinity both for the compound 5, derived from 3, and for 22, the direct analog of 4, while the same modification with an isopinocampheyl (7, 23) and adamantyl group (10, 24) in both substitutions produce a loss of CB2R-affinity. In contrast, the cyclohexyl carboxamide derived 17 has 8.5-fold higher affinity towards CB2Rs as compared to the parent compound 3, but the same group in 24 lead to a 4.6-fold decrease in CB2R-affinity respect to 4.
Overall, as previously reported, the benzocyclopentapyrazole core showed preference for CB2Rs. The introduction of a chlorine as well as its exchange with the methyl group in all new hybrid compounds seems to play a modest role in lowering the levels of CB2-affinity as compared to the leads 1 and 2. Nevertheless, these compounds provide further information regarding the structural features responsible for CB2 affinity and selectivity.
INTRINSIC ACTIVITY THROUGH IN VITRO ASSAYS
According to their highest affinity towards CB2 receptors, compounds 8, 17 and 22 were assayed to determine their intrinsic activity and, consequently, their potential therapeutic application.
In a first step, tests were carried out to determine eventual agonist activity of the compounds by treating HL-60 cells and determining increasing of P-ERK ½ expression by western blot analysis compare to the case of vehicle. Within this step, studies concerning time-related induction of P-ERK ½ expression were also performed, these experiments indicating 15 minutes as better exposure time of HL-60 cells to the cannabinoid ligands (data not shown). For all the assayed compounds, as for the reference cannabinoid agonist WIN 55,212-2, a significant increase of P-ERK ½ expression was highlighted in HL-60 treated cells (Fig. 3). A maximum effect was detected for WIN 55,212-2 at the dose of 75 nM (dose response curve not shown, percentage of increment versus vehicle: +83.2 ± 18.9 %), while the assayed new CB2 ligands elicited the highest enhancement of P-ERK ½ expression compare to the vehicle at the doses of 125 nM for compounds 8 and 17 (+54.5 ± 12.1 %, and +82.5 ± 19.1 %, respectively), and of 50 nM for 22 (+46.4 ± 15.6 %).
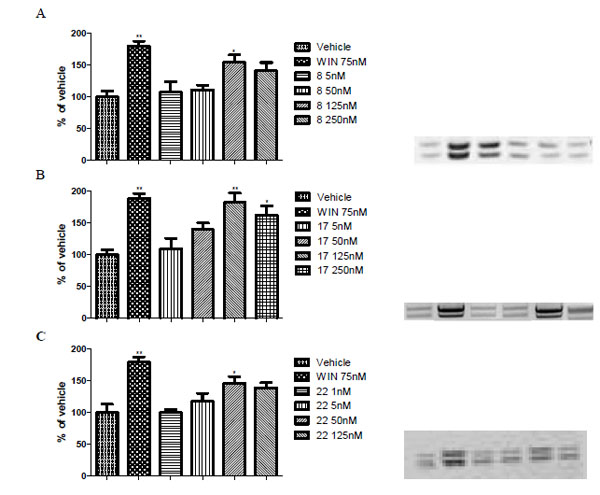
Panels A-C show dose response study of P-ERK 1/2 expression following a 15 min exposure to cannabinoid receptor agonist WIN55, 212-2 (75 nM; WIN) or to compounds 8 (Panel A), 17 (Panel B), and 22 (Panel C) at different concentrations. On the right side are shown representative western blots of P-ERK expression. Data are expressed as mean percentage of vehicle ± SEM and are the results of five separate experiments. *p < 0.05 vs vehicle; **p < 0.05 vs vehicle.
Final experiments were performed for all the derivatives in order to verify the correspondence between the detected effect and CB2 modulation. The approach was to repeat the experiments as in the first step (doses corresponding to the maximum effect) but by exposing the cells to the reference CB2 antagonist AM630 (75 nM) 5 minutes before the treatment with the compounds to be tested. As reported in Fig. (4), the effect on P-ERK ½ expression due to both the new compounds and WIN 55,212-2 was counteracted by the action of the reference CB2 antagonist.

Panels A-C show the inhibition by the CB2 selective antagonist AM630 (AM) of the effect of CB2 agonists towards HL-60 cells: 8 (Panel A), 17 (Panel B), and 22 (Panel C). The concentrations of the CB2 agonists were defined according to the peaking doses detected in Figure 3; that of AM630 was chosen among the doses lacking an intrinsic activity and providing an appropriate block of the CB2 receptor. The assays were carried out with 5 min pre-treatment with AM630, followed by a 15 min exposure to CB2 agonist derivatives. On the right side are shown representative western blots of P-ERK expression. Data are expressed as mean percentage of vehicle ± SEM and are the results of five separate experiments. *p < 0.05 vs vehicle; **p < 0.05 vs vehicle; #p < 0.05 vs WIN55,212-2; §p < 0.05 vs 8, 17, or 22.
According to the detected behavior from in vitro assays, CB2 agonist activity was then highlighted for compounds 8, 17 and 22, confirming previous data concerning analogue CB2 selective 1,4-dihydroindeno[1,2-c]pyrazole derivatives [45].
CONCLUSIONS
In summary, we have used molecular hybridization of existing CB2 ligands to efficiently create novel CB2-selective 1,4-dihydroindeno[1,2-c]pyrazoles. By utilizing the results of SAR studies around 5, we prepared compound 22 with nanomolar affinity for CB2R and 195.6-fold selectivity for CB2 over CB1Rs.
According to previous reported data concerning analogue tricyclic pyrazole derivatives [45], CB2 agonism activity has been preliminary determined by in vitro assays for these new compounds.
These results confirm the effect of the flat tricyclic core of 1,4-dihydroindeno[1,2-c]pyrazoles compared to the not condensed analogues (i.e. SR144528) to assure agonist rather than antagonist activity towards CB2R. Moreover this study further support the development of new potential chemical entities for CB2 mediated pharmacology therapies based on pyrazole based tricyclic condensed scaffold, i.e. for neuropathic and/or inflammatory pain treatments [48].
EXPERIMENTAL SECTION
General Procedures
Melting points were obtained on a Köfler melting point apparatus and are uncorrected. IR spectra were recorded as nujol mulls on NaCl plates with a Perkin Elmer 781 IR spectrophotometer and are expressed in ν (cm-1). All NMR spectra were taken on a Varian XL-200 NMR spectrometer with 1H and 13C being observed at 200 and 50 MHz respectively. Chemical shifts for 1H and 13C NMR spectra were reported in δ or ppm downfield from TMS [(CH3)4Si]. Multiplicities are recorded as s (singlet), br s (broad singlet), d (doublet), br d (broad doublet), dd (doublet of doublets), m (multiplet). Atmospheric Pressure Ionization Electrospray (API-ES) mass spectra were obtained on a Agilent 1100 series LC/MSD spectrometer. Compound purity was assessed by elemental analysis, on a Perkin-Elmer 240-B analyzer, for C, H, and N; measured percent values were within ± 0.4% of theoretical ones. All the tested compounds possessed a purity >95%. All reactions involving air or moisture-sensitive compounds were performed under argon atmosphere. Flash chromatography was performed using pre-packed Biotage® SNAP silica-gel cartridges. Thin layer chromatography (TLC) was performed with Polygram SIL N-HR/HV254 pre-coated plastic sheets (0.2 mm). The amines for the synthesis of final compounds were purchased by Sigma-Aldrich: fenchylamine was synthesized according to the literature procedure [49].
Syntheses of α-keto-γ-hydroxyesters 28 and 29, tricyclic esters 30 and 31, carboxylic acids 32 and 33, and compounds 3, 4 and 17 were previously described in our recent paper [45].
General procedure I:
Synthesis of Carboxamides 5-16, 19, 22-25and Carbohydrazides 20, 21. A mixture of the dihydroindeno [1,2-c] pyrazole-3-carboxylic acid (32, 33) (1 eq, 0.51 mmol), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide (EDC) (1.2 eq) and 1-hydroxybenzotriazole (BtOH) (1.2 eq) in dichloromethane (2.36 mL) was stirred at room temperature for 0.5 h. A solution of the requisite amine or hydrazine (1.02 eq) in dichloromethane (1.6 mL) was dropwise added and the whole was stirred at room temperature for 22 h. The solution was concentrated under reduced pressure and the analytically pure product was isolated after purification by flash chromatography.
N-Fenchyl-7-chloro-1-(2,4-dichlorophenyl)-6-methyl-1,4-dihydroindeno[1,2-c]pyrazole-3-carboxamide (5).
General procedure I was used to convert 32 and fenchylamine into the title product. The purification by flash chromatography (petroleum ether/Et2O 9:1) afforded 5 (86%) as a yellow solid. Rf = 0.46 (petroleum ether/Et2O 9:1); mp 210-212 °C; IR 3380 (NH), 1680 (CO); 1H NMR [200 MHz CDCl3]: δ = 0.88 (s, 3H), 1.12 (s, 3H), 1.19 (s, 3H), 1.23-1.83 (m, 7H), 2.41 (s, 3H), 3.79 (br s, 1H), 3.84 (s, 2H), 6.96 (s, 1H), 7.01 (d, NH exch. with D2O, J = 9.0 Hz), 7.43 (s, 1H), 7.48 (dd, 1H, Jo = 8.2 Hz, Jm = 2.0 Hz), 7.57 (d, 1H, J = 8.4 Hz), 7.68 (d, 1H, J = 2.0 Hz); 13C NMR [50 MHz CDCl3]: δ = 19.77 (CH3), 20.42 (CH3), 21.31(CH3), 25.97 (CH3), 27.32 (CH2), 29.34 (CH2), 30.94 (CH2), 39.52 (CH), 42.72 (C), 48.15 (CH2), 48.64 (C), 63.17 (CH), 119.44 (CH), 128.23 (CH), 128.56 (CH), 129.61 (CH), 130.41 (C), 130.62 (CH), 131.69 (C), 132.66 (C), 134.59 (C), 135.82 (C), 136.02 (C), 142.12 (C), 148.09 (C x 2), 150.66 (C), 162.27 (CO); API-ES calcd for C28H28Cl3N3O: 528.90.57, found: 529.20.
N-Bornyl-7-chloro-1-(2,4-dichlorophenyl)-6-methyl-1,4-dihydroindeno[1,2-c]pyrazole-3-carboxamide (6).
General procedure I was used to convert 32 and bornylamine into the title product. The purification by flash chromatography (petroleum ether/acetone 9:1) afforded 6 (82%) as a yellow solid. Rf = 0.42 (petroleum ether/acetone 9:1); mp 234-235 °C; IR 3280 (NH), 1650 (CO); 1H NMR [200 MHz CDCl3]: δ = 0.91 (s, 6H), 1.00 (s, 3H), 1.16-1.35 (m, 3H), 1.57 (s, 1H), 1.60-1.70 (m, 3H), 2.41 (s, 3H), 3.83 (s, 2H), 4.43-4.50 (m, 1H), 6.90-7.00 (m, 2H), 7.42 (s, 1H), 7.49 (d, 1H, J = 8.6 Hz), 7.57 (d, 1H, J = 8.6 Hz), 7.69 (s, 1H); 13C NMR [50 MHz CDCl3]: δ= 13.85 (CH3), 18.65 (CH3), 19.86 (CH3), 20.41 (CH3), 28.07 (CH2), 28.39 (CH2), 29.34 (CH2), 37.45 (CH), 44.96 (C), 48.22 (CH2), 49.73 (C), 53.60 (CH), 119.44 (CH), 128.29 (CH), 128.57 (CH), 129.70 (CH), 130.41 (C), 130.60 (CH), 131.69 (C), 132.66 (C), 134.59 (C), 135.82 (C), 136.02 (C), 142.12 (C), 148.10 (C x 2), 150.66 (C), 161.81 (CO); API-ES calcd for C28H28Cl3N3O: 528.90, found: 529.15.
N-Isopinocampheyl-7-chloro-1-(2,4-dichlorophenyl)-6-methyl-1,4-dihydroindeno[1,2-c]pyrazole-3-carboxamide (7).
General procedure I was used to convert 32 and isopinocampheylamine into the title product. The purification by flash chromatography (petroleum ether/EtOAc 9:1) afforded 7 (89%) as a yellow solid. Rf = 0.40 (petroleum ether/EtOAc 9:1); mp 200-201 °C; IR 3395 (NH), 1685 (CO); 1H NMR [200 MHz CDCl3]: δ = 1.11-1.25 (m, 9H), 1.69-2.00 (m, 5H), 2.41 (s, 3H), 2.55-2.80 (m, 2H), 3.85 (s, 2H), 4.40-4.62 (m, 1H), 6.83 (d, 1H, NH exch. with D2O, J = 9.0 Hz), 6.94 (s, 1H), 7.43 (s, 1H), 7.48 (dd, 1H, Jo = 8.6 Hz, Jm = 2.0 Hz), 7.56 (d, 1H, J = 8.2 Hz), 7.68 (d, 1H, J = 2.0 Hz); 13C NMR [50 MHz CDCl3]: δ = 20.41 (CH3), 20.84 (CH3), 23.36 (CH3), 28.06 (CH3), 29.36 (CH2), 35.24 (CH2), 37.02 (CH2), 38.52 (C), 41.62 (CH), 45.91 (CH2), 47.62 (CH), 47.83 (CH), 119.44 (CH), 128.34 (CH), 128.58 (CH), 128.79 (C), 129.66 (CH), 130.32 (C), 130.62 (CH), 131.78 (C), 132.70 (C), 134.70 (C), 135.72 (C), 136.24 (C), 142.22 (C), 148.11 (C x 2), 162.17 (CO); API-ES calcd for C28H28Cl3N3O: 528.90, found 528.10.
N-Myrtanyl-7-chloro-1-(2,4-dichlorophenyl)-6-methy-l-1,4-dihydroindeno[1,2-c]pyrazole-3-carboxamide (8).
General procedure I was used to convert 32 and cis-myrtanylamine into the title product. The purification by flash chromatography (petroleum ether/EtOAc 9:1) afforded 8 (78%) as a yellowish solid. Rf = 0.26 (petroleum ether/EtOAc 9:1); mp 230-231 °C; IR 3378 (NH), 1683 (CO); 1H NMR [200 MHz CDCl3]: δ = 0.90-1.70 (m, 11H), 1.75-2.20 (m, 4H), 2.41 (s, 3H), 3.30-3.63 (m, 2H), 3.84 (s, 2H), 3.87-4.00 (m, 1H), 6.92-7.00 (m, 2H, NH, exch. with D2O), 7.40-7.62 (m, 3H), 7.68 (d, 1H, J = 1.8 Hz); 13C NMR [50 MHz CDCl3]: δ = 19.85 (CH3), 20.42 (CH3), 23.22 (CH3), 26.02 (CH2), 27.98 (CH2), 29.35 (CH2), 33.28 (CH2), 38.72 (C), 41.36 (CH), 41.52 (CH), 43.80 (CH2), 44.74 (CH), 119.43 (CH), 128.31 (CH), 128.57 (CH), 129.57 (CH), 130.32 (C), 130.66 (CH), 131.79 (C), 132.71 (C), 134.71 (C), 135.73 (C), 136.22 (C), 141.55 (C), 142.14 (C), 148.09 (C x 2), 161.67 (CO); API-ES calcd for C28H28Cl3N3O: 528.90, found 529.15.
N-Menthyl-7-chloro-1-(2,4-Dichlorophenyl)-6-methyl-1,4-dihydroindeno[1,2-c]pyrazole-3-carboxamide (9).
General procedure I was used to convert 32 and menthylamine into the title product. The purification by flash chromatography (petroleum ether/acetone 9:1) afforded 9 (70%) as a yellow solid. Rf = 0.64 (petroleum ether/acetone 9:1); mp 185-186 °C; IR 3278 (NH), 1618 (CO); 1H-NMR [200 MHz CDCl3]: δ = 0.70-1.09 (m, 9H), 1.40-1.81 (m, 6H), 1.87-2.20 (m, 2H), 2.41 (s, 3H), 3.85 (s, 2H), 3.90-4.00 (m, 1H), 6.63 (br d, NH exch. with D2O, J = 9.8 Hz), 6.94 (s, 1H), 7.43 (s, 1H), 7.45-7.62 (m, 3H), 7.68 (s, 1H); 13C-NMR [50 MHz CDCl3]: δ = 16.23 (CH3), 20.42 (CH3), 21.17 (CH3), 22.16 (CH3), 23.88 (CH2), 26.82 (CH), 29.40 (CH2), 31.99 (CH2), 34.54 (CH2), 43.12 (CH), 47.94 (CH), 49.78 (CH), 119.44 (CH), 128.33 (CH), 128.57 (CH), 128.79 (C), 129.63 (CH), 130.35 (C), 130.65 (CH), 131.77 (C), 132.67 (C), 134.66 (C), 135.73 (C), 136.20 (C), 141.54 (C), 148.13 (C x 2), 160.91 (CO); API-ES calcd for C28H30Cl3N3O: 530.92, found 531.25.
N-Adamant-1-yl-7-chloro-1-(2,4-dichlorophenyl)-6-methyl-1,4-dihydroindeno[1,2-c]pyrazole-3-carboxamide (10).
General procedure I was used to convert 32 and 1-adamantylamine into the title product. The purification by flash chromatography (petroleum ether/acetone 9.5:0.5) afforded 10 (97%) as a yellow solid. Rf = 0.27 (petroleum ether/acetone 9.5:0.5); mp 254.8-255.2 °C; IR 3278 (NH), 1633 (CO); 1H-NMR [200 MHz CDCl3]: δ = 1.63-1.88 (m, 6H), 1.99-2.12 (m, 9H), 2.40 (s, 3H), 3.84 (s, 2H), 6.68 (br s, NH exch. with D2O), 6.93 (s, 1H), 7.42 (s, 1H), 7.45-7.58 (m, 2H), 7.68 (s, 1H); 13C-NMR [50 MHz CDCl3]: δ = 20.45 (CH3), 29.37 (CH2), 29.49 (CH2 x 3), 36.39 (CH x 3), 41.70 (CH2 x 3), 52.03 (C), 119.43 (CH), 128.29 (CH), 128.57 (CH), 129.67 (CH), 130.38 (C), 130.56 (CH), 131.75 (C), 132.67 (C), 134.61 (C), 135.76 (C), 136.15 (C), 142.97 (C), 148.15 (C x 2), 150.77 (C), 160.91 (CO); API-ES calcd for C28H26Cl3N3O: 526.88, found 527.10.
N-Adamant-2-yl-7-chloro-1-(2,4-dichlorophenyl)-6-methyl-1,4-dihydroindeno[1,2-c]pyrazole-3-carboxamide (11).
General procedure I was used to convert 32 and 2-adamantylamine hydrochloride into the title product. Because of an excess of the hydrochloride salt, an amount of two equiv of TEA was used in this reaction. The purification by flash chromatography (petroleum ether/EtOAc 8.5:1.5) afforded 11 (78%) as a yellow solid. Rf = 0.44 (petroleum ether/Et2O 8.5:1.5); mp 276-277 °C; IR 3300 (NH), 1630 (CO); 1H-NMR [200 MHz CDCl3]: δ = 1.50-2.15 (m, 14H), 2.41 (s, 3H), 3.83 (s, 2H), 4.17-4.38 (m, 1H), 6.95 (s, 1H), 7.20-7.36 (m, NH exch. with D2O), 7.43 (s, 1H), 7.48 (dd, 1H, Jo = 8.4 Hz, Jm = 2.2 Hz), 7.56 (d, 1H, J = 8.2 Hz), 7.68 (s, 1H); 13C-NMR [50 MHz CDCl3]: δ= 20.45 (CH3), 27.16 (CH), 27.25 (CH), 29.33 (CH2), 29.70 (CH), 31.97 (CH2), 32.05 (CH2 x 2), 37.16 (CH2 x 2), 37.54 (CH), 53.15 (C), 119.44 (CH), 128.25 (CH), 128.55 (CH), 128.63 (C), 129.70 (CH), 130.42 (C), 130.58 (CH), 131.74 (C), 132.68 (C), 134.61 (C), 135.81 (C), 136.10 (C), 142.32 (C), 148.09 (C x 2), 160.90 (CO); API-ES calcd for C28H26Cl3N3O: 526.88, found 527.20.
N-1-Adamantanmethyl-7-chloro-1-(2,4-dichlorophenyl)-6-methyl-1,4-dihydroindeno[1,2-c]pyrazole-3-carboxamide (12).
General procedure I was used to convert 32 and 1-adamantylamine into the title product. The purification by flash chromatography (petroleum ether/EtOAc 9:1) afforded 12 (58%) as a yellow solid. Rf = 0.35 (petroleum ether/EtOAc 9:1); mp 248-250 °C; IR 3252 (NH), 1643 (CO); 1H-NMR [200 MHz CDCl3]: δ= 1.32-1.80 (m, 12H), 1.95-2.10 (m, 3H), 2.41 (s, 3H), 3.14 (d, 2H, J = 6.7 Hz), 3.84 (s, 2H), 6.95 (s, 1H), 6.97-7.03 (m, NH exch. with D2O), 7.43 (s, 1H), 7.48 (dd, 1H, Jo = 8.6 Hz, Jm = 2.2 Hz), 7.56 (d, 1H, J = 8.6 Hz), 7.68 (d, 1H, J = 1.8 Hz); 13C-NMR [50 MHz CDCl3]: δ= 20.42 (CH3), 29.24 (CH x 3), 29.35 (CH2), 29.69 (CH2), 36.91 (CH2 x 3), 40.27 (CH2 x 3), 50.65 (CH2), 119.42 (CH), 128.29 (CH), 128.57 (CH), 129.60 (CH), 130.34 (C), 130.64 (CH), 131.76 (C), 132.68 (C), 134.66 (C), 135.76 (C), 136.16 (C), 142.11 (C), 148.09 (C x 2), 150.74 (C), 160.90 (CO); API-ES calcd for C29H28Cl3N3O: 540.91, found 541.20.
N-(1-Cyclohexylethyl)-7-chloro-1-(2,4-dichloropheny-l)-6-methyl-1,4-dihydroindeno[1,2-c]pyrazole-3-carboxamide (13).
General procedure I was used to convert 32 and S-(+)-cyclohexylethylamine into the title product. The purification by flash chromatography (petroleum ether/acetone 9:1) afforded 13 (20%) as a yellow solid. Rf = 0.49 (petroleum ether/acetone 9:1); mp 225-227 °C; IR 3305 (NH), 1646 (CO); 1H-NMR [200 MHz CDCl3]: δ = 1.10-1.35 (m, 7H), 1.60-1.95 (m, 7H), 2.41 (s, 3H), 3.85 (s, 2H), 3.98-4.15 (m, 1H), 6.67 (br d, NH, exch. with D2O, J = 9.0 Hz ), 6.94 (s, 1H), 7.43 (s, 1H), 7.48 (dd, 1H, Jo = 8.4 Hz, Jm = 1.8 Hz), 7.56 (d, 1H, J = 8.6 Hz), 7.68 (d, 1H, J = 2.0 Hz); 13C-NMR [50 MHz CDCl3]: δ= 18.01 (CH3), 20.41 (CH3), 26.18 (CH2 x 2), 26.39 (CH2), 29.17 (CH2 x 2), 29.36 (CH2), 43.24 (CH), 49.28 (CH), 119.44 (CH), 128.30 (CH), 128.56 (CH), 128.68 (C), 129.64 (CH), 130.35 (C), 130.62 (CH), 131.75 (C), 132.68 (C), 134.65 (C), 135.76 (C), 136.18 (C), 142.24 (C), 148.1o (C), 150.74 (C), 161.02 (CO); API-ES calcd for C26H26Cl3N3O: 502.86, found 503.15.
Trans-N-(4-Methylcyclohexyl)-7-chloro-1-(2,4-dichlorophenyl)-6-meth-yl-1,4-dihydroindeno[1,2-c]pyrazole-3-carboxamide (14) and cis-N-(4-Methylcyclohexyl)-7-chloro-1-(2,4-dichloroph-enyl)-6-methyl-1,4-dihydroindeno[1,2-c]pyrazole-3-carboxa-mide (15).
General procedure I was used to convert 32 and 4-methylcyclohexylamine into the title products. The purification by flash chromatography (petroleum ether/EtOAc 9:1) afforded isomer trans 14 (60%) as a yellow solid. Rf = 0.19 (petroleum ether/EtOAc 9:1); mp 218-219 °C; IR 3303 (NH), 1630 (CO); 1H-NMR [200 MHz CDCl3]: δ = 0.94 (d, 3H, J = 6.0 Hz), 1.05-1.30 (m, 5H), 1.65-1.80 (m, 2H), 2.00-2.18 (m, 2H), 2.41 (s, 3H), 3.84 (s, 2H), 4.10-4.27 (m, 1H), 6.76 (br s, 1H, NH, exch. with D2O), 6.94 (s, 1H), 7.43 (s, 1H), 7.48-7.56 (m, 2H), 7.68 (m, 1H); 13C-NMR [50 MHz CDCl3]: δ = 20.44 (CH3), 22.20 (CH3), 29.35 (CH2), 31.99 (CH), 33.15 (CH2 x 2), 33.90 (CH2 x 2), 48.38 (CH), 119.45 (CH), 128.33 (CH), 128.58 (CH), 128.68 (C), 129.63 (CH), 130.34 (C), 130.64 (CH), 131.80 (C), 132.69 (C), 134.71 (C), 135.41 (C), 136.24 (C), 141.56 (C), 142.28 (C), 148.11 (C), 160.86 (CO); API-ES calcd for C25H24Cl3N3O: 488.84, found 490.20; and isomer cis15 (20%) as a yellow solid. Rf = 0.27 (petroleum ether/EtOAc 9:1); mp 203-205 °C; IR 3301 (NH), 1623 (CO); 1H-NMR [200 MHz CDCl3]: δ = 0.93 (d, 3H, J = 6.0 Hz), 1.48-2.00 (m, 9H), 2.41 (s, 3H), 3.84 (s, 2H), 4.10-4.27 (m, 1H), 6.95 (s, 1H), 7.00 (d, 1H, NH, exch. with D2O, J = 7.4 Hz), 7.43 (s, 1H), 7.48 (dd, 1H, Jo = 8.4 Hz, Jm = 2.2 Hz), 7.56 (d, 1H, J = 8.2 Hz), 7.69 (d, 1H, J = 2.0 Hz); 13C-NMR [50 MHz CDCl3]: δ = 20.41 (CH3), 20.97 (CH3), 29.32 (CH2 x 2), 29.69 (CH), 30.05 (CH2 x 2), 30.39 (CH2), 45.40 (CH), 119.43 (CH), 128.28 (CH), 128.55 (CH), 129.65 (CH), 130.46 (C), 130.61 (CH), 131.76 (C), 132.67 (C), 134.64 (C), 135.77 (C), 136.15 (C), 142.28 (C), 148.09 (C x 2), 150.70 (C), 160.94 (CO); API-ES calcd for C25H24Cl3N3O: 488.84, found 490.20.
N-Cycloheptyl-7-chloro-1-(2,4-dichlorophenyl)-6-methyl-1,4-dihydroindeno[1,2-c]pyrazole-3-carboxamide (16).
General procedure I was used to convert 32 and cycloheptylamine into the title product. The purification by flash chromatography (petroleum ether/Et2O 6:4) afforded 16 (72%) as a yellow solid. Rf = 0.47 (petroleum ether/Et2O 6:4); mp 227-229 °C; IR 3323 (NH), 1652 (CO); 1H-NMR [200 MHz CDCl3]: δ = 1.34-1.80 (m, 10H), 1.87-2.19 (m, 2H), 2.41 (s, 3H), 3.84 (s, 2H), 4.00-4.28 (m, 1H), 6.86 (br d, NH exch. with D2O, J = 8.2 Hz), 6.94 (s, 1H), 7.43 (s, 1H), 7.47 (dd, 1H, Jo = 8.6 Hz, Jm = 2.0 Hz), 7.55 (d, 1H, J = 8.6 Hz), 7.68 (d, 1H, J = 2.2 Hz); 13C-NMR [50 MHz CDCl3]: δ = 20.42 (CH3), 24.25 (CH2 x 2), 28.06 (CH2 x 2), 29.34 (CH2), 35.12 (CH2 x 2), 50.34 (CH), 119.43 (CH), 128.31 (CH), 128.58 (CH), 129.26 (C), 129.63 (CH), 130.36 (C), 130.63 (CH), 131.79 (C), 132.70 (C), 134.68 (C), 135.76 (C), 136.20 (C), 142.31 (C), 148.10 (C x2), 160.55 (CO); API-ES calcd for C25H24Cl3N3O: 488.84, found 489.25.
N-(1-(1-Naphthyl)ethyl)-7-chloro-1-(2,4-dichlorophenyl)-6-methyl-1,4-dihydroindeno[1,2-c]pyrazole-3-carboxamide (19).
General procedure I was used to convert 32 and 1-(1-naphthyl)ethylamine into the title product. The purification by flash chromatography (petroleum ether/EtOAc 8.5:1.5) afforded 19 (86%) as a yellowish solid. Rf = 0.48 (petroleum ether/EtOAc 8.5:1.5); mp 188-189 °C; IR 3250 (NH), 1640 (CO); 1H-NMR [200 MHz CDCl3]: δ = 1.78 (d, 3H, J = 6.6 Hz), 2.41 (s, 3H), 3.86 (s, 2H), 6.00-6.28 (m, 1H), 6.93 (s, 1H), 7.22 (d, NH exch. with D2O, J = 8.8 Hz), 7.40-7.65 (m, 8H), 7.75-7.90 (m, 2H), 8.24 (d, 1H, J = 8.4 Hz); 13C-NMR [50 MHz CDCl3]: δ = 20.36 (CH3), 21.12 (CH3), 29.30 (CH2), 44.36 (CH), 119.39 (CH), 122.61 (CH), 123.32 (CH), 125.20 (CH), 125.68 (CH), 126.44 (CH), 128.20 (CH x 2), 128.50 (CH), 128.70 (CH), 129.54 (CH), 130.21 (C), 130.48 (CH), 131.02 (C), 131.57 (C), 132.66 (C), 133.84 (C), 134.67 (C), 135.55 (C), 136.14 (C), 138.31 (C), 141.78 (C), 147.97 (C x 2), 150.75 (C), 160.6 (CO); API-ES calcd for C30H22Cl3N3O: 546.87, found 547.20.
N-(Pyrrolidin-1-yl)-7-chloro-1-(2,4-dichlorophenyl)-6-methyl-1,4-dihydroindeno[1,2-c]pyrazole-3-carbohydrazide (20).
General procedure I was used to convert 32 and N-aminopyrrolidine hydrochloride into the title product. Because of an excess of the hydrochloride salt, an amount of two equiv of TEA was used in this reaction. The purification by flash chromatography (petroleum ether/EtOAc 4:6) afforded 20 (77%) as a yellowish solid. Rf = 0.21 (petroleum ether/EtOAc 4:6); mp 207-208 °C; IR 3380 (NH), 1680 (CO); 1H-NMR [200 MHz CDCl3]: δ = 1.82-2.00 (m, 4H), 2.41 (s, 3H), 2.95-3.10 (m, 4H), 3.86 (s, 2H), 6.94 (s, 1H), 7.42 (s, 1H), 7.45-7.53 (m, 2H), 7.68 (d, 1H, J = 1.6 Hz); 13C-NMR [50 MHz CDCl3]: δ = 20.42 (CH3), 22.33 (CH2 x 2), 29.35 (CH2), 55.40 (CH2 x 2), 119.45 (CH), 128.32 (CH), 128.56 (CH), 129.02 (C), 129.57 (CH), 130.16 (C), 130.63 (CH), 131.75 (C), 132.74 (C), 134.81 (C), 135.62 (C), 136.29 (C), 141.25 (C), 148.06 (C x 2), 159.89 (CO); API-ES calcd for C22H19Cl3N4O: 461.77, found 462.05.
N-(Homopiperidin-1-yl)-7-chloro-1-(2,4-dichlorophenyl)-6-methyl-1,4-dihydroindeno[1,2-c]pyrazole-3-carbohydrazide (21).
General procedure I was used to convert 32 and N-aminohomopiperidine into the title product. The purification by flash chromatography (petroleum ether/EtOAc 6:4) afforded 21 (64%) as a yellow solid. Rf = 0.51 (petroleum ether/EtOAc 6:4); mp 219-220 °C; IR 3346 (NH), 1670 (CO); 1H-NMR [200 MHz CDCl3]: δ = 1.60-1.83 (m, 8H), 2.41 (s, 3H), 3.13-3.21 (m, 4H), 3.84 (s, 2H), 6.94 (s, 1H), 7.42 (s, 1H), 7.47 (dd, 1H, Jo = 8.6 Hz, Jm = 2.0 Hz), 7.54 (d, 1H, J = 8.2 Hz), 7.68 (d, 1H, J = 1.6 Hz), 8.10 (br s, NH exch. with D2O); 13C-NMR [50 MHz CDCl3]: δ = 20.43 (CH3), 26.31 (CH2 x 2), 27.02 (CH2 x 2), 29.32 (CH2), 58.29 (CH2 x 2), 119.46 (CH), 128.31 (CH), 128.57 (CH), 129.09 (C), 129.56 (CH), 130.23 (C), 130.65 (CH), 131.73 (C), 132.73 (C), 134.77 (C), 135.68 (C), 136.23 (C), 141.48 (C), 148.10 (C x 2), 159.33 (CO); API-ES calcd for C24H23Cl3N4O: 489.82, found 490.05.
N-Fenchyl-6-chloro-1-(2,4-dichlorophenyl)-7-methyl-1,4-dihydroindeno[1,2-c]pyrazole-3-carboxamide (22).
General procedure I was used to convert 33 and fenchylamine into the title product. The purification by flash chromatography (petroleum ether/Et2O 8:2) afforded 22 (86%) as a yellow solid. Rf = 0.40 (petroleum ether/Et2O 8:2); mp 210-212 °C; IR 3380 (NH), 1680 (CO); 1H-NMR [200 MHz CDCl3]: δ = 0.88 (s, 3H), 1.12 (s, 3H), 1.19 (s, 3H), 1.19-1.90 (m, 7H), 2.34 (s, 3H), 3.79 (s, 1H), 3.85 (s, 2H), 6.83 (s, 1H), 7.01 (br d, NH exch. with D2O, J = 9.8 Hz), 7.46-7.60 (m, 3H), 7.68 (s, 1H); 13C-NMR [50 MHz CDCl3]: δ = 19.74 (CH3), 20.34 (CH3), 21.27 (CH3), 25.95 (CH3), 27.29 (CH2), 29.29 (CH2), 30.91 (CH2), 39.48 (CH), 42.69 (C), 48.13 (CH2), 48.60 (C), 63.14 (CH), 120.77 (CH), 126.86 (CH), 128.14 (CH), 128.30 (C), 129.69 (CH), 129.98 (C), 130.50 (CH), 131.78 (C), 133.03 (C), 134.36 (C), 135.93 (C), 142.11 (C), 148.44 (C x 2), 150.86 (C), 162.23 (CO); API-ES calcd for C28H28Cl3N3O: 528.90, found 529.20.
N-Isopinocampheyl-6-chloro-1-(2,4-dichlorophenyl)-7-methyl-1,4-dihydroindeno[1,2-c]pyrazole-3-carboxamide (23).
General procedure I was used to convert 33 and isopinocampheylamine into the title product. The purification by flash chromatography (petroleum ether/EtOAc 9:1) afforded 23 (78%) as a yellow solid. Rf = 0.44 (petroleum ether/EtOAc 9:1); mp 208-209 °C; IR 3247 (NH), 1695 (CO); 1H-NMR [200 MHz CDCl3]: δ = 1.11-1.25 (m, 9H), 1.69-2.00 (m, 5H), 2.34 (s, 3H), 2.57-2.80 (m, 2H), 3.85 (s, 2H), 4.37-4.64 (m, 1H), 6.65-6.92 (m, 2H), 7.38-7.77 (m, 4H); 13C-NMR [50 MHz CDCl3]: δ = 20.29 (CH3), 20.79 (CH3), 23.31 (CH3), 28.01 (CH3), 29.28 (CH2), 35.17 (CH2), 36.96 (CH2), 38.45 (C), 41.56 (CH), 45.85 (CH2), 47.53 (CH), 47.78 (CH), 120.74 (CH), 126.85 (CH), 128.22 (CH), 128.58 (C), 129.71 (CH), 129.87 (C), 130.45 (CH), 131.81 (C), 133.10 (C), 134.37 (C), 135.79 (C), 136.09 (C), 142.18 (C), 148.43 (C), 150.92 (C), 161.07 (CO); API-ES calcd for C28H28Cl3N3O: 528.90, found 529.05.
N-Adamant-1-yl-6-chloro-1-(2,4-dichlorophenyl)-7-methyl-1,4-dihydroindeno[1,2-c]pyrazole-3-carboxamide (24).
General procedure I was used to convert 33 and 1-adamantylamine into the title product. The purification by flash chromatography (petroleum ether/EtOAc 9:1) afforded 24 (90%) as a yellow solid. Rf = 0.52 (petroleum ether/EtOAc 9:1); mp 245-247.2 °C; IR 3248 (NH), 1621 (CO); 1H-NMR [200 MHz CDCl3]: δ = 1.45-1.85 (m, 6H), 1.95-2.30 (m, 9H), 2.33 (s, 3H), 3.83 (s, 2H), 6.67 (br s, NH exch. with D2O), 6.80 (s, 1H), 7.39-7.59 (m, 3H), 7.67 (d, 1H, J = 2.0 Hz); 13C-NMR [50 MHz CDCl3]: δ = 19.72 (CH3), 28.71 (CH2), 28.85 (CH2 x 3), 35.74 (CH x 3), 41.05 (CH2 x 3), 51.39 (C), 120.15 (CH), 126.28 (CH), 127.61 (CH), 127.83 (C), 129.14 (CH), 129.33 (C), 129.86 (CH), 131.25 (C), 132.46 (C), 133.77 (C), 135.23 (C), 135.44 (C), 142.33 (C), 147.91 (C), 150.36 (C), 160.25 (CO); API-ES calcd for C28H26Cl3N3O: 526.88, found 527.10.
N-(1-Cyclohexyl)-7-chloro-(2,4-dichlorophenyl)-6-methyl-1,4-dihydroindeno[1,2-c]pyrazole-3-carboxamide (25).
General procedure I was used to convert 33 and cyclohexylamine into the title product. The purification by flash chromatography (petroleum ether/EtOAc 9:1) afforded 25 (87%) as a white solid. Rf = 0.26 (petroleum ether/EtOAc 9:1); mp 240-243 °C; IR 3287 (NH), 1683 (CO); 1H-NMR [200 MHz CDCl3]: δ = 1.25-2.20 (m, 10H), 2.34 (s, 3H), 3.85 (s, 2H), 3.92-4.10 (m, 1H), 6.72-6.85 (m, 2H, NH exch. with D2O), 7.41-7.61 (m, 3H), 7.68 (d, 1H, J = 2.0 Hz); 13C-NMR [50 MHz CDCl3]: δ = 20.37 (CH3), 24.97 (CH2 x 2), 25.57 (CH2), 29.34 (CH2), 33.17 (CH2 x 2), 48.11 (CH), 120.79 (CH), 126.92 (CH), 128.28 (CH), 128.55 (C), 129.73 (CH), 129.93 (C), 130.54 (CH), 131.89 (C), 133.14 (C), 134.45 (C), 135.86 (C), 136.14 (C), 142.29 (C), 148.49 (C), 151.96 (C), 160.76 (CO); API-ES calcd for C24H22Cl3N3O: 474.81, found 475.10.
N-(Naphthalen-1-yl)-7-chloro-1-(2,4-dichlorophenyl)-6-methyl-1,4-dihydroindeno[1,2-c]pyrazole-3-carboxamide (18).
A mixture of acid 32 (0.51 mmol, 1 eq) and SOCl2 (3 eq) was refluxed in toluene (5 mL) for 3 h. After evaporation at reduced pressure of the SOCl2 in excess, the residue was dissolved in CH2Cl2 (3 mL) and the resulting solution was added to a solution of 1-naphthylamine in CH2Cl2 (3 mL), while keeping the temperature at 0 °C. The mixture was stirred at room temperature overnight, then the solution was poured into H2O and brine and extracted with CHCl3. The organic phase was dried (Na2SO4) and concentrated: the purification of the residue by flash chromatography (petroleum ether/EtOAc 9:1) afforded 18 (34%) as a brown solid. Rf = 0.50 (petroleum ether/EtOAc 9:1); mp 176-178 °C; IR 3250 (NH), 1640 (CO); 1H-NMR [200 MHz CDCl3]: δ = 2.34 (s, 3H), 3.92 (s, 2H), 7.00 (s, 1H), 7.28-8.06 (m, 10H), 8.29 (d, 1H, J = 7.6 Hz), 9.31 (br s, NH, exch. with D2O); 13C-NMR [50 MHz CDCl3]: δ = 20.47 (CH3), 29.39 (CH2), 119.40 (CH), 119.56 (CH), 120.49 (CH), 125.25 (CH), 125.87 (CH), 125.97 (CH), 126.24 (CH), 128.41 (CH), 128.65 (CH), 128.76 (CH), 128.92 (C), 129.58 (CH), 130.25 (C), 130.77 (CH), 130.87 (C), 131.81 (C), 132.09 (C), 132.85 (C), 134.08 (C), 134.97 (C), 135.69 (C), 136.40 (C), 142.13 (C), 148.05 (C x 2), 159.94 (CO); API-ES calcd for C28H18Cl3N3O: 518.82, found 519.15.
RADIORECEPTOR BINDING ASSAYS
Male CD-1 mice (Charles River, Calco, LC, Italy), weighing 30-35 g, were used. Mice were housed in plastic cages under a 12 h artificial light-dark cycle (lights off at 8.00 p.m.), at a constant temperature (22 ± 2 °C). All experimental procedures were performed in strict accordance with the E.C. regulation for care and use of experimental animals (EEC No. 86/609). Animals were maintained with water and rodent chow ad libitum. Where not specified, solvents and reagents were obtained from Sigma Aldrich, Milano, Italy.
For the assays, mice were killed by cervical dislocation and the brain (minus cerebellum) and spleen were rapidly removed and placed on an ice-cold plate. After thawing, tissues were homogenated in 20 vol. (wt/v) of ice-cold TME buffer (50 mM Tris-HCl, 1 mM EDTA and 3.0 mM MgCl2, pH 7.4). The homogenates were centrifuged at 1,086 x g for 10 min at 4 °C, and the resulting supernatants were centrifuged at 45,000 x g for 30 min.
[3H]-CP-55,940 binding was performed by the method previously described [45]. Briefly, the membranes (30-80 µg of protein) were incubated with 0.5-1.0 nM of [3H]-CP-55,940 (specific activity 180 Ci/mmol; New England Nuclear, Boston, MA) for 1 h at 30 °C in a final volume of 0.5 ml of TME buffer containing 5 mg/ml of fatty acid-free bovine serum albumin. Non-specific binding was estimated in the presence of 1 µM of CP-55,940 (Tocris, Bristol, U.K.). All binding studies were performed in disposable glass tubes pre-treated with Sigma-Cote (Sigma Chemical Co. Ltd., Poole, UK), in order to reduce non-specific binding. The reaction was terminated by rapid filtration through Whatman GF/C filters presoaked in 0.5% polyethyleneimine (PEI) using a Brandell 36-sample harvester (Gaithersburg, MD, USA). Filters were washed five times with 4 ml aliquots of ice cold Tris HCl buffer (pH 7.4) containing 1 mg/mL BSA The filter bound radioactivity was measured in a liquid scintillation counter (Tricarb 2900, Packard, Meridien, USA) with 4 ml of scintillation fluid (Ultima Gold MV, Packard).
Protein determination was performed by means of Bradford protein assay using BSA as a standard according to the protocol of the supplier (Bio-Rad, Milan, Italy) [50].
All experiments were performed in triplicate and results were confirmed in at least five independent experiments. The results concerning the new compounds were compared to those of the reference CB2 selective derivative SR144528 (Sanofi-Synthélabo, now Sanofi-Aventis). Data from radioligand inhibition experiments were analyzed by nonlinear regression analysis of a Sigmoid Curve using Graph Pad Prism program. IC50 values were derived from the calculated curves and converted to Ki values as previously described [51].
IN VITRO CB2 ACTIVITY EVALUATION
Where not specified, solvents and reagents were obtained from Sigma Aldrich, Milano, Italy. Human promyelocytic leukemia HL-60 cells from the European Collection of Cell Cultures (ECACC, Salisbury, UK) were purchased from Sigma-Aldrich (Milano, Italy). Cell lines were grown at 37 °C in humidified 5% CO2 in RPMI 1640 medium (Gibco-BRL, Gaithersburg, MD, USA) supplemented with 10% heat-inactivated fetal bovine serum FBS (Gibco-BRL), 25 mM HEPES, 2.5 mM sodium pyruvate, 20 µg/ml gentamicin. Culture medium was added every two days and experiments were made at 80% cell confluency. Tested and reference compounds were dissolved in culture medium with 1% DMSO and treatments were made for time course, dose response and competition studies in a volume of 10 µl/ml of cell suspension.
In order to verify whether the observed effects were CB2 receptor specific, a 5 min pre-treatment with the CB2 receptor antagonist AM630 (75 nM) was performed before the exposure to the compounds that displayed a significant induction of P-ERK (as shown by western blot analysis). AM630 was purchased from Tocris (Bristol, U.K.). The dose of AM630 was chosen among the doses lacking an intrinsic activity and providing an appropriate block of the CB2R.
For western blot analysis after appropriate time of exposure, cells were collected by centrifugation at 1000Xg and the resulting pellets were washed in ice-cold PBS buffer by centrifugation at 1000X g. The cells were then lysed at 4 °C in 50 μl of 20 mM HEPES buffer (pH 7.9) containing: NaCl 125 mM; MgCl2 5 mM; glycerol 12%; ethylenediaminetetracetic acid (EDTA) 0.2 mM; Nonidet P-40 0.1%; dithithreitol (DTT) 5 mM; phenilmethylsulphonil fluoride (PMSF) 0.5 mM; leupeptin 0.5 μg/mL; and pepstatin A 0.7 μg/ml. The extracts were then centrifuged at 10000 g (at 4 °C) for 15 min and the resulting supernatant was collected as total cell extracts. An aliquot was analysed for protein concentration determination by using Protein assay kit II (Bio-Rad Laboratories, Hercules, CA, USA), and the rest was frozen at –80 °C until assayed. Western blot studies were performed as previously described [45]. Immunoreactive bands were visualized with a Fuji Las 1000 image analyzer (Raytest Isotopenmessgeräte GmbH, Straubenhartd, Germany). The optical density of immunoreactive bands was measured using a specific software (AIDA 2.11, Raytest Isotopenmessgeräte GmbH, Straubenhartd, Germany). One-way ANOVA was performed as a statistical analysis using Graph Pad Prism program (San Diego, CA, USA).
CONFLICT OF INTEREST
None declared.
ACKNOWLEDGEMENTS
Ministero dell’Istruzione, dell’Università e della Ricerca (MIUR) and Regione Autonoma della Sardegna (RAS) are acknowledged for economic support (MIUR: project "Pirazol-Cannabinoidi”, DM 10262; RAS: project “CB2 cannabinoidi”).

