All published articles of this journal are available on ScienceDirect.

Identification of G-Quadruplex Inducers Usinga Simple, Inexpensiveand Rapid High Throughput Assay, and TheirInhibition of Human Telomerase
Abstract
Telomeres are protein and DNA complexes located atchromosome ends. Telomeric DNA is composed of a double stranded region of repetitive DNA followed by single-stranded 3' extension of aG-rich sequence. Single-stranded G-rich sequencescan fold into G-quadruplex structures,and molecules that stabilize G-quadruplexes are known to inhibit the enzyme telomerase and disrupt telomere maintenance. Because telomere maintenance is required for proliferation of cancer cells, G-quadruplex stabilizers have become attractive prospects for anticancer drug discovery.However, telomere-targeting G-quadruplex ligands have yet to enter the clinic owing in part to poor pharmacokinetics and target selectivity. Increasing the pharmacophore diversity of G-quadruplex and specifically telomeric-DNA targeting agents should assist in overcoming these shortcomings. In this work, we report the identification and validation ofligands that bind telomeric DNA and induce G-quadruplex formationusing the NCI Diversity Set I, providing validation of anextremely simple, rapid and high-throughput screen using FRET technology. Hits from the screen were validated by examining telomerase inhibition and G-quadruplex inductionusing CD spectroscopy and DNA polymerase stop assays. We show that two known DNA binding molecules, ellipticine derivativeNSC 176327 (apyridocarbazole) and NSC 305831 (an antiparasitic hetero-cyclediamidine referred to as furamidine and DB75),are selective induceG-quadruplex formation in the human telomeric sequence and bind telomeric DNA quadruplexes in the absence of stabilizing monovalent cations with molar ratios(molecule: DNA)of 4:1and 1.5:1, respectively.
1. INTRODUCTION
The telomere is a nucleoprotein complex located at the ends of eukaryotic chromosomesand is essential for genome integrity. For much of the cell cycle, telomeric DNA is presumably maintained in a t-loop structure, which serves to protect vulnerable chromosome ends [1, 2]. Many of the key proteins that function at the telomere have been identified, although their interplay is still imperfectly understood and structural data are only available for a few [3]. Telomeric DNA is comprised of one strand of simple guanine-rich repeats and a complementary C-rich strand, 5'-dTTAGGG/3'dAATCCC in mammals, for most of its length, culminating in a short single-strand overhang of the G-rich sequence at the extreme 3' end.Single-stranded oligonucleotides composed of telomeric G-rich repeats are known to fold into a wide variety of four-stranded quadruplex structures, giving rise to speculation telomeric DNA itself can fold into G-quadruplex structures [4].
Normal DNA polymerases cannot fully replicate the ends of telomeric DNA [5]. This"end-replication problem" combined with end processing and degradationresults in the ero-sion of telomeric DNA at a rate of approximately 50–100 bases per cell cycle in human cells. In the absence of any compensating mechanism,this telomere shorteningleads to a DNA damage response at the telomere culminating in cell cycle arrest or cell death [6]. Cellscan compensate for the end-replication problem through the action of the reverse transcriptase telomerase. Telomerase is a unique reverse transcriptase that binds to the guanine-rich 3' end of telomeric DNA and synthesizes the addition of hexanucleotide repeats onto the end using its own integral RNA template. Germ-line cells have significant telomerase activity and maintain telomere length even after prolonged cycling making them essentially immortal.By contrast, adult stem cells are mortal and have some telomerase activity, but not enough to replenish all telomeres lost from telomere erosion [7]. Fully differentiated somatic cells have little or no telomerase activity and cannot undergo prolonged cellular proliferation. In contrast to somatic cells, the telomeres of tumor cells do not shorten during proliferation but instead tend to have short yet stable telomeres. In most cases, this stabilization is accomplished by telomerase, which is expressed in 80–85% of human tumors even though it is absent in neighboring normal somatic tissue. Telomerase thus plays a key role in maintaining the malignant phenotype by stabilizing telomere length and integrity, a core hallmark of cellular immortalization and cancer [8].The roles of telomerase in ensuring cellular immortality and its differential expression in cancer cells compared to normal cells has made telomerase an important research focus and validated anticancer drug target [9-12].
G-quadruplexes are higher-order DNA and RNA structures formed from G-rich sequences that are built around tetrads of hydrogen-bonded guanine bases. Potential quadruplex sequences have been identified in G-rich eukaryotic telomeres and in non-telomeric genomic DNA. The natural role and biological validation of these structures is in being explored, and there is particular interest in them as targets for therapeutic intervention [13]. The in vivo importance of G-quadruplex-DNA has been speculated for quite some time [14]. A growing body of evidence for the biological relevance of G-quadruplex-DNA has emerged from recent literature, and putative G-quadruplex forming sequences are thoroughly distributed along the human genome (37,000 sequences) [15]. These sequences are particularly enriched at telomeric regions and gene promoters [16]. The putative quadruplex formation in promoter regions correlates with gene expression, and several proteins with diverse functions have been shown to interact specifically with G-quadruplexes [17].As a result of these potentially diverse G-quadruplex locations, inducing G-quadruplex formation with small molecules has the potential to affect several biological pathways[18]. In particular, the formation of G-quadruplex-DNA in a telomeric primer has been reported to obstruct telomerase associationand inhibit primer extension [19]. Additionally, G-quadruplex formation caninhibit DNA binding by telomere binding proteins in vitro and induce genomic instability by displacing telomere-binding proteins such as POT1 and TRF2in cells [20-22]. Because G-quadruplexes inhibit telomerase and have other effects on telomere structure and maintenance they are considered effective leads for the development of anti-cancer drug development, and there is a general consensus that G-quadruplex ligands could lead to the discovery of novel anti-cancer agents [23].
Telomere-binding G-quadruplex ligands have yet to reach the clinic, though some have shown reasonable abilities to reduce tumor size in mouse models [20, 24, 25]. Possible reasons for the lack of clinical success include a lack of specificity leading to nonspecific effects and poor tumor penetrating ability.Most G-quadruplex-binding ligands contain large polyaromatic surfaces with several cationic charges, which may contribute to their nonspecific affects. In addition, their commonly high charge density likely hinders cellular uptake, as demonstrated for BRACO19 [26]. By comparison, one G-quadruplex ligand, quarfloxin (CX-3543),which appears to disrupt nucleolin/rDNA quadruplex complexes, is in clinical trials [27]. Using quarfloxin, which contains only a single cationic charge, as a guide suggests that decreasing the cationic content of G-quadruplex-binding ligands will improve the chances of cellular penetration.
Given the large number of polycationicchromophores already explored, we reasoned that a new approach towards the identification of telomere-binding agents with enhanced selectivity and good cellular penetration is merited. Here, we describe our efforts thatestablish a simple, cost-effective high throughput-compatible assay for the identification of ligands that bind telomeric DNA and induce quadruplex formation based on Förster resonance energy transfer (FRET). We validated the assay using the NCI Diversity SetI and confirmed the activity of several hits using appropriately designed biochemical assays.
2. RESULTS
In previous reports, fluorescence-based assays havebeen used to identify molecules that stabilize G-quadruplexes by measuring the effect on the thermal stability of the folded structures [28, 29]. Here, we tested a simplified and alternative approach using an assay that identifies molecules that promote G-quadruplexes under buffer conditions that disfavor their formation by lacking stabilizing monovalent cations such as Na+ or K+ [13]. The assay platform is based onthe ligand stimulated folding of a commercially available oligonucleotide containing 3.5telomeric repeats, fluoresceine-d(GGGTTA)3GGG-tetramethylrhodamine (Fam-21hT-Tam), into a G-quadruplex structure. Folding brings the extremities of the oligonucleotide into close proximity leading toFRET between the donorfluoresceine and the acceptor tetramethylrhodamine. As a consequence, G-quadruplex formation can be measured by simultaneously monitoring the quenching of fluoresceine at 550 nM and FRET-induced fluorescence at595nMto determine the extent of G-quadruplex formation [28].
We optimized the assay conditionsusing potassium as the inducer of G-quadruplex formation, since it is known that the presence of a monovalent ion stabilizes G-quadruplex structures [30]. The optimized assay included 40 nMFam-21hT-Tam in TRIS-EDTA (pH 7.5) in the absence of additionalcations. Test ligand, KCl in validation assays,and Fam-21hT-Tam were incubated for 5 min before fluorescence was measured. Comparison of no ligand (negative control) and ligand (50 mMKCl) produced a Z-factor of0.88 for readings at 595 and 0.77 at 550 nm. Further, low plate-to-plate and day-to-day variabilityand no sensitivity to DMSO up to 10% was observed, verifying the robust quality of the assay.
2.1. Primary screen for the identification of G-quadruplex inducers using a FRET assay
To test the ability of a G-quadruplex formation assay to identify biologically active compounds, we first screened the 1,990 compounds contained in theNCI Diversity Set I. When reading the plates at 550 nm, a low signal (10,000-15,000 relative fluorescence units)indicated quenching of the donor emission and formation of the G-quadruplex structure in the oligonucleotide, and a high signal at 550 nm (35,000-50,000 relative fluorescence units)indicated that the oligonucleotide wasunstructured. Compounds from the NCI Diversity Set I were preliminarily classified asG-quadruplex inducers if fluorescence at 550 nmwas <20,000. The screen revealed 55 compounds as potential G-quadruplex stabilizing compounds (2.8% hit rate). The hits from the diversity screen were retested with pure compounds and only 29 induced a decreased 550 nm signal <20,000 indicativeof G-quadruplex formation.Of the 29 hits from the duplicate assay, we selected the 16 most promising compounds based on synthetic tractability and drug likeness for titration experiments based on structures and reported biochemical and biological profiles of the compounds. Titration of these 16 compounds in the G-quadruplex formation assay revealed 10 potent G-quadruplex inducers. Potency was defined as the ability to induce half-maximum G-quadruplex formation at < 10 µM compound. Purified samples of NSC 12155, 17600, 35489, 95609, 130813, 176327, 305831, 354961, 357777, and 638432, were obtained from the NCI repository for further testing Fig. (1). Notably, NSC 176327 was also identified as a stabilizer of the G-quadruplex formed by the MYC promoter sequence using a G-quadruplex stability screen [31].
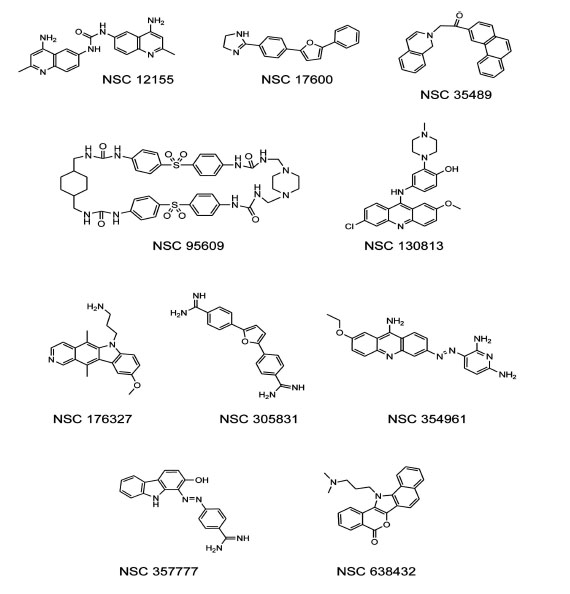
Structures of human telomere G-quadruplex inducers identified in the NCI diversity set I.
2.2. G-quadruplex inducers inhibit telomerase
To evaluate the effect of the identified G-quadruplex inducers on telomerase we utilized a direct primer extension assay of recombinant telomerase overexpressed in HEK293T/17 cells. Compounds were tested at 50 μM in an initial screen Fig. (2). A 2'-O-Me oligomer,5’-CAGUUAGGGUUAG (hTR-AS), was used as a positive control for telomerase inhibition [32]. Compounds NSC 12155, 176327, 305831, 354961, and 35489 were chosen for further investigation because they showed 60% or greaterinhibition of telomerase activity at 50 µM (93, 97, 86, 69, and 63 %, respectively, Fig. (2). The concentration dependence of telomerase inhibition for these compounds was determined as described in Fig. (3). The IC50calculated were 36, 1.5, 7.5, 18, and 65μM, respectively for the five NSC 12155, 176327, 305831, 354961, and 35489.
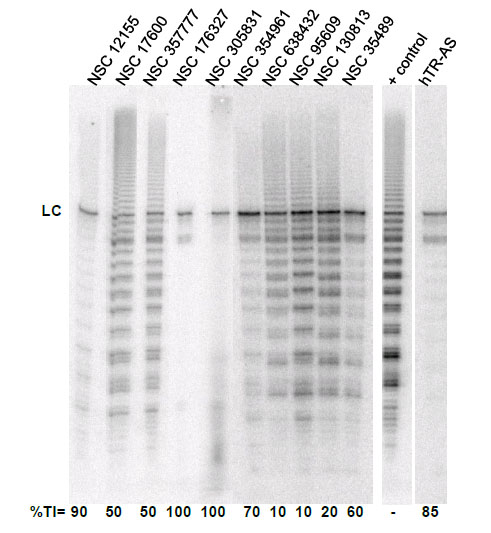
Telomerase inhibition by G-quadruplex inducersidentified in the NCI diversity set I. The ability of compounds identified as G-quadruplex inducers to inhibit human telomerase was tested at 10 µM compound using a direct primer extension assay. Percent inhibition is reported below each lane. Above each lane is the compound tested in the experiment. The lane marked + control is a positive control for telomerase activity in the absence of inhibitor; and the lane marked hTR-AS is acontrol for telomerase inhibition in the presence of a telomerase inhibitorhTR-AS (200nM) that acts as a template agonist. LC (loading and recovery control) is a 115 nucleotide, 5´-32P-end labeled DNA oligonucleotide. %TI, Percent inhibition of telomerase activity compared to the positive control.
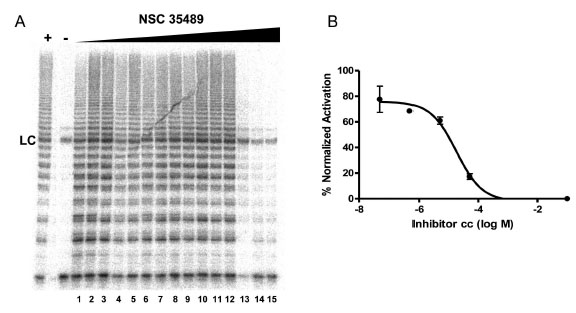
Concentration dependence of telomerase inhibition by NSC 35489.A. Inhibition of telomerase by NSC 35489. Lanes 1-3, 5 nM; lanes 4-6, 50 nM; lanes 7-9, 500 nM; lanes 10-12, 5 μM; and lanes 13-15, 50 μM. The lane marked – is a control for telomerase activity in the presence of a known telomerase inhibitor: hTR-AS at 200nM. LC is a loading control as in Figure 2. B. Dose response curve for telomerase inhibition by NSC 35489.
2.3. G-quadruplex ligand specificity revealed by polymerase stop assay
Compounds that bind to and stabilize G-quadruplex DNA commonly exhibit nonspecific DNA-binding properties.To assess extent of nonspecific binding, we utilized a Taq polymerase stop assay. Taq polymerase activity was measured using two types of DNA templates: one contained four repeats of the human telomeric sequence, Temp [TTAGGG]4 [33], which contains a G-quadruplex forming sequence; and a second template, Temp [TTAGAG]4, which contains four repeats of a non-G-quadruplex-forming sequence. The latter served as a control for nonspecific inhibitory activity. G-quadruplex stabilizers cause a polymerase stop product at the G-quadruplex-forming site.Inhibition of primer extension using Temp [TTAGAG]4 and stop products seen before the G-quadruplex pause site when using Temp[TTAGGG]4 indicate nonspecific inhibition. Presumably nonspecific inhibition results from either ligand binding to the primer/template duplex or other unrevealed mechanisms of inhibition. In either case, nonspecific inhibition is evidenced by inhibition of primer extension without specific G-quadruplex stops [34].
Compounds NSC354961, NSC 305831, NSC 12155, NSC 35489, and NSC 176327 were evaluatedin the polymerase stop assay. Fig. (4) shows the results of Taq DNA polymerase primer extension on the DNA templates. BRACO19, a known G-quadruplex stabilizing ligand, was used as a positive control for G-quadruplex formation. NSC 176327, NSC 12155, NSC 354961, and NSC 305831 showed polymerase stop products at the G-quadruplex forming site indicative of G-quadruplex formation. To further evaluate the ability to selective induce G-quadruplex formation, the dose-dependence on the stop assay was evaluated. We found that only NSC 176327 and NSC 305831 showed specific G-quadruplex stop site products, suggesting that these compounds inhibited the DNA synthesis of Temp [TTAGGG]4 by Taq polymerase by selectivelyinitiated G-quadruplex formation in the template Fig. (5). At higher concentration, nonspecific inhibition was evidenced by the inhibition of full-length product formation concomitant with the increase of unextended primer. Primer extension reactions using the non-G-quadruplex-forming Temp [TTAGAG]4revealed no G-quadruplex stop products for these two compounds at < 5 µM. The remainder of the compounds showed stop products consistent with nonspecific DNA binding (data not shown). These results encouraged us to further investigate the G-quadruplex structures inducedby NSC 176327 and NSC 305831.
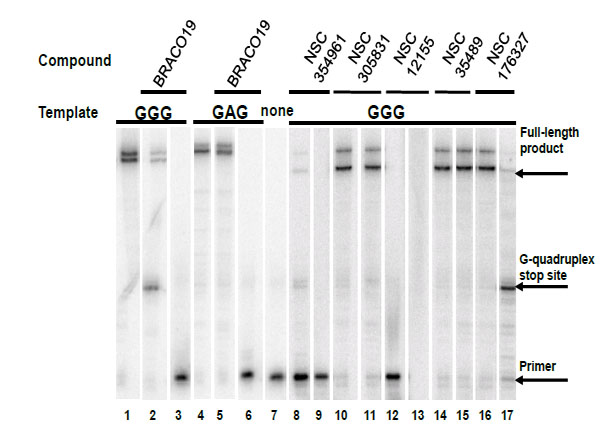
Selectivity test of hits for G-quadruplex stabilization by polymerase stop assay. Compounds were tested in a Taq polymerase stop assay to evaluate specificity towards induction of G-quadruplex compared to inhibition of Taq by other mechanisms. Lane 1: Temp [TTAGGG]4, G-quadruplex forming template as control. Lane 2: Temp [TTAGGG]4 with 1 µM BRACO19. Lane 3: Temp [TTAGGG]4 with 50 µM BRACO19. Lane 4: Temp [TTAGAG]4, non G-quadruplex forming template as control. Lane 5: Temp [TTAGAG]4 with 1 µM BRACO19. Lane 6: Temp [TTAGAG]4 with 50 µM BRACO19. Lane 7: Labeled primer (P) at 15 nM with no template added. Lanes 8-9: NSC354961 at 5 and 50 µM. Lanes 10-11: NSC 305831 at 0.5 and 5 µM. Lanes 12-13: NSC 12155 at 5 and 50 µM. Lanes 14-15: NSC 35489 at 5 and 50 µM. Lanes 16-17: NSC 176327 at 0.5 and 5 µM.
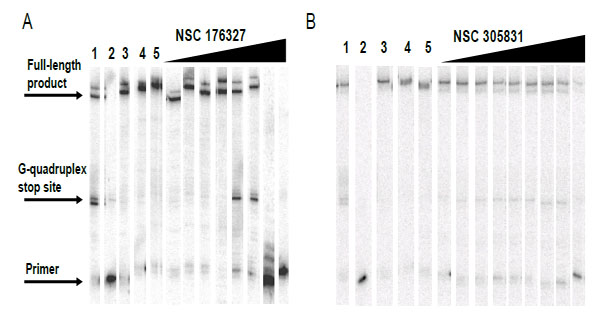
Concentration dependent inhibition of DNA synthesis by NSC 176327 and NSC 305831in a polymerase stop reaction using the human telomeric sequence [TTAGGG]4.A. Lane 1, Temp [TTAGGG]4 with 1 µM BRACO19;lane 2, labeled primer at 15 nM, lane 3; Temp [TTAGGG]4,lane 4; Temp [TTAGAG]4;and lane 5, Temp [TTAGAG]4 with NSC 176327 at 5 µM. The concentrations of NSC 176327 were 0.1, 0.25, 0.5, 2.5, 5, 10, 25, and 50 μM. B. Lane 1, Temp [TTAGGG]4 with 1 µM BRACO19;lane 2, labeled primer at 15 nM, lane 3; Temp [TTAGGG]4,lane 4; Temp [TTAGAG]4;and lane 5, Temp [TTAGAG]4 with NSC305831 at 5 µM.The concentrations of NSC 305831 were 10, 15, 20, 22.5, 25, 27.5, 30, 35, and 40 μM.
2.4. Evaluation of G-quadruplex induction by circular dichroism
Circular dichroismcan be used to determine binding mode and stoichiometry of ligand-DNA interactions. Ligand-DNA interactions can be studied by virtue of the interpretation of ligand-induced changes in CD signals resulting from the coupling of electric transition moments of the ligand and DNA bases within the asymmetric DNA environment [35, 36]. CD spectra of G-quadruplex structures are distinct from other DNA structuresallowing the observation of a ligand-induced change in CD spectrum of a DNA sample to be used as evidence of ligand-induced structure changes. [36] Because the various folding topologies of G-quadruplexes have diagnostic CD spectra, CD can assist in determining the folding topology induced by G-quadruplex ligands [37].
The interactions of compounds NSC 176327 and 305831 with human telomeric oligonucleotide d[5’-G3(T2AG3)3-3’], hTelo4, which contains four repeats of human telomeric DNA, were monitored by CD spectroscopy. Titrations of ligands into a fixed concentration of hTelo4 were conducted to determine the stoichiometry of the binding as well as characterize the G-quadruplex structure. The spectrum of hTelo4 alone showed a major positive band at 256 nm, and a minor band at 295 nm indicative of unfolded, G-rich DNA Fig. (6) [21, 37]. The titration of increasing amounts of NSC 176327 (0.5 to 6 mol equivalents) with a fixed concentration of human telomeric DNA revealed a major negative peak at 253 nm and the disappearance of the positive peak at 295nm present before Fig. (6A). This suggests the ligand-induced formation of a new structure, presumably a G-quadruplex, although it is not clear what conformation is favored owing to the significant contribution of the chromophore to the spectra. This steep decrease of the 253 nm band stopped when a 4:1 ratio of NSC 176327 to DNA was reached consistent with saturation of binding Fig. (6C).Titration of NSC305831 resulted in a new spectrum with a significant increase in the 293 nm peak and a small positive peak at 283 nm. This profile is similar to the one seen in a K+ solution, suggesting the formation of a mixed-type or 3+1 type structure. This positive band increased until a 1.5:1 ratio of NSC 305831 to DNA was reached Figs. (6B and 6D).
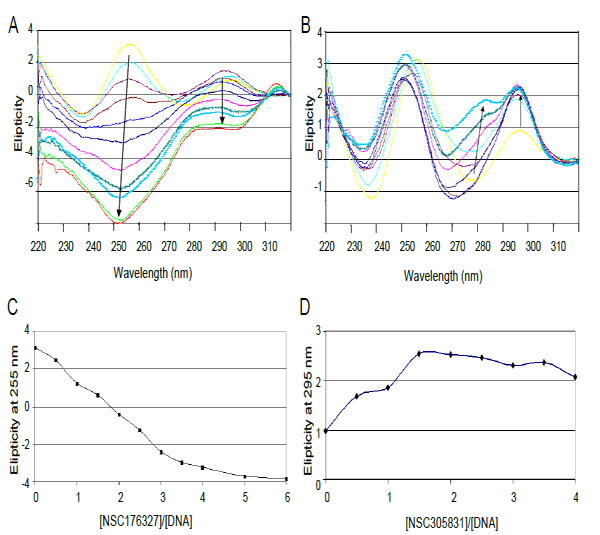
CD spectra of titration of d[5'-G3(T2AG3)3] with (A) NSC 176327 and (B) NSC 305831.A. CD spectra of NSC 176327 titration. The spectrum in yellow is DNA without ligand. Arrows indicate increasing ligand concentration B. CD spectra of NSC 305831 titration. Colored as for A. C. Plot of molar ellipticity at 255 nm versus NSC 176327-DNA molar ratio. The spectrum equilibrates at a ratio of 4:1. D. Plot of molar ellipticity at 295 nm versus NSC 305831-DNA molar ratio. The spectrum equilibrates at a ratio of 1.5:1.
3. DISCUSSION
Here we demonstrate that a simple, inexpensive, and rapid G-quadruplex formation assay can accurately identify G-quadruplex binding compounds that inhibit telomerase. The assay represents a significant departure of other high throughput assays used to identify G-quadruplex interacting compounds by focusing on the propensity to induce G-quadruplex formation as opposed to stabilize G-quadruplex structures [28, 29]. The thermal stability assay requires 2-3 hrs per plate for the thermal melt compared to 5 min for G-quadruplex formation in our assay. As proof of principle, our assay identified several G-quadruplex inducers including confirmation thatNSC 176327 and NSC 305831 are selective G-quadruplex inducers of the human telomeric sequencethat inhibit human telomerase see Fig. (1) for compound structures). NSC 305831 appears to induce the formation of mixed-type, or 3+1, structure with an apparent binding stoichiometry of 1.5:1. Finding NSC 305831 in our screen is not surprising as analogs of NSC 305831 have been reported as G-quadruplex ligands [35]. Owing to its ability to bind varieties of DNA structures, NSC 305831 exhibits a variety of biochemical activities including inhibition of tyrosyl-DNA phosphodiesterase [38], to which we can firmly add telomerase inhibition. We could not assess the structure induced by NSC 176327 as it produced a complex spectrum dominated by the spectral changes in the ligand itself. However, we determined that NSC 176327 bound with a 4:1 molar ratio.NSC 176327 is an analog of the plant alkaloid ellipticine, and it is reported to bind to DNA with high affinity (106 M-1) [39-42]. There is reference to ellipticine as a telomerase inhibitor through G-quadruplex binding, though no primary data have been published [43]. We have strengthened this point by directly demonstrating that the ellipticine analog NSC 176327inhibits telomerase and induced G-quadruplex formation as indicated by the Taq polymerase stop assay.NSC 176327 was also identified as a ligand for the G-quadruplex formed by the MYCpromoter and a derivative of NSC 176327, NSC86374, was found to display increased selectivity for G-quadruplex DNA compared to double stranded DNA as well as selectivity for the MYC promoter G-quadruplex compared to the telomeric G-quadruplex suggesting that the ellipticine scaffold may prove highly useful for engineering compounds with high G-quadruplex selectivity .
G-quadruplexes present challenges with respect to drug discovery owing to their low abundance compared to duplex DNA and the varieties of G-quadruplexes that may exist within chromosomal DNA. In addition, the highly charged nature of most current telomere-targeting ligands decreases their ability to penetrate solid tumors. Here, we show that a simple G-quadruplex formation assay can identify new scaffolds upon which to begin developing more potent and selective G-quadruplex-binding ligands that are inhibitors of telomerase. The assay represents a generalizable platform that could include other nucleic acid structures and, importantly, has proved successful in identifying active ligands characterized bystructural diversity and identifiedtelomerase inhibitors that bind telomeric DNA and induce G-quadruplex formation even in the absence of monocations.
4. MATERIALS AND METHODS
4.1. Materials
All DNAs were purchased from IDT (Coralville, IA) and purified by polyacrylamide gel electrophoresis. The dual-labeled oligonucleotide Fam-21hTelo-Tam (5'-FAM-GGGTTAGGGTTAGGGTTAGGG-TAMRA) was obtained from IDT and was purified by HPLC. The NCI diversity set I, a library of 1,990 chemically and biologically diverse compounds was obtained from the NCI/DTP Open Chemical Repository (http://dtp.cancer.gov). Compounds were obtained as powders, dissolved in DMSO to a concentration of 10 mM, and daughter plates were prepared by dilution to 1.25 mM in DMSO for use in subsequent assays. Positive hits were validated from compounds obtained in pure form from the NCI.hTERT- and hTR- expressing plasmids, pVan107 and pBS-U1-hTR, respectively, were a gift from Dr. Joaquim Lingner [44].
4.2. G-quadruplex formation assay and screen of the NCI Diversity Set I.
G-quadruplex formation assays were conducted in 96-well plates. Each well contained 40 nMof Fam-21hTelo-Tam inTEbuffer (10 mMTRIS, 1 mMEDTA, pH 7.5). Compounds from the NCI Diversity Set I were added to a final concentration of 10 μM. Controls included Fam-21hTelo-Tam in the presence or absence of 50 mMKCl. Samples were incubated in a total volume of 100 μL for 5 minat ambient temperature before fluorescence readings were obtained. Samples were excited at 485 nm and read at 550 nm and 595 nm on a HTS-7000 Plus BioAssay Reader (Perkin-Elmer). The initial hits from the primary screen were tested in duplicate. Positives were titrated to generate dose-response curves using 40 nM, 500 nM, 1000 nM, 2000 nM, and 4000 nM concentrations.
4.3. Super-telomerase Extract
Super-telomerase cell extracts were prepared as reported by Cristofari et al. [44]. In short, HEK293T/17 cell (2-6x105 per well in a 6-well plate) were transfected with 4 μg of total plasmid DNA using Lipofectamine 2000 (Invitrogen) following the manufacturer’s protocol. The mass ratio of hTERT- and hTR- expressing plasmids was 1:5 (0.75 μgpVan107 and 3.38 μgpBS-U1-hTR). 24 h post transfection, cells were trypsinized, transferred to a 25 cm2 flask, and grown a subsequent 24 h. Two days post transfection, cells (3-4x106) were detached with trypsin, washed once in PBS, and lysed in 400 µl of Chaps lysis buffer (10mMTris-HCl, pH 7.5, 1mMMgCl2, 1mMEGTA, 0.5% CHAPS, 10% glycerol, protease inhibitor cocktail (Roche), and 5mM β-mercaptoethanol). After incubation at 4ºC for 30 min on a rotator, cell debris was removed by centrifuging extracts at 4ºC for 10 min at 13,000 × g. Protein concentration of extracts was determined using the Coomassie Plus Assay kit (Pierce). The supernatant was aliquoted in portions of 4 µl, quick frozen on dry ice, and stored without loss of activity for several months at -80 ºC.
4.4. Telomerase Assay
Telomerase activity was measured using a modification of a previously described direct primer-extension assay [45]. Each 25 µl reaction contained 50 mMTris-HCl, pH 8.0, 50 mMKCl, 1 mMMgCl2, 5 mM β-mercaptoethanol, 1 mMspermidine, 1 µM human telomere primer (5'-TTAGGGTTAGGGTTAGGG), 0.5 mMdATP, 0.5 mMdTTP, 2.9 µM dGTP, 0.17 µM [α-32P]-dGTP (3000 Ci/mmol, 10 μCi/μl; Perkin-Elmer), and 4 µl of the super-telomerase cell extract (1.6μg total protein /μl). Primer extension reactions were carried out at 30 °C for 90 min. The known telomerase inhibitor,hTR-AS (a 2-O'-Methyl RNA, 5'-CAGUUAGGGUUAG), was used as a positive inhibitor control. After the addition of a 32P-labeled loading control (15 or 115 nucleotide, 5’-end labeled DNA oligonucleotide, 1000 cpm per reaction), the primer extension products were extracted with phenol/chloroform/isoamyl alcohol and ethanol precipitated in the presence of 0.6 M NH4OAc and 35 ng/μl glycogen. Products were precipitated at -80 °C in 2.5 vol of absolute ethanol for 30 min followed by centrifugation at 22,000gat 4 °C for 25 min and washed twice with 70% ethanol. The final pellets were dissolved in a formamide loading buffer containing 40% formamide, 10 mMTris-HCl, pH 8.0, 10 mMEDTA, 0.05% xylene cyanol, and 0.05% bromophenol blue. The products were heated at 95 °C for 5 min and resolved on a prewarmed, 0.4 mm thick, 20 x 20 cm, 10% polyacrylamide/7 M urea/1 ×TBE gel. Gels were run at 800 V for 45 min in 1 ×TBE. Gels weredried and exposed to a phosphorimager screen (Molecular Dynamics) overnight, imaged using a phosphorimager (Molecular Dynamics Storm 860), and quantified with Image Quant (version 5.2). The intensities of each band in each sample were summed and normalized to the loading control.
IC50for the most potent compounds were determined by direct telomerase assay with five point dose response curves for each compound (50 µM, 5 µM, 0.5 µM, 50 nM, and 5 nM). Each reaction was run in triplicate. Assay controls included buffer (positive) and 200 nMhTR-AS (negative). Each kinetic trace was normalized to the loading control signal to correct for loading of the extracts. Data was analyzed using Graph Pad Prism 5 for Windows to obtain dose response curves and the IC50 values.
4.5. Polymerase Stop Assay
The specificity of G-quadruplex binders was characterized using a modification of a previously reported polymerase stop assay[21].Single-stranded oligonucleotides were 5'-end labeled using T4 polynucleotide kinase and [γ-32P] ATP at 37ºC. The kinase activity was inactivated by heating at 70ºC for 8 min and the labeled primer was purified on a Microspin G-25 column (GE Healthcare). Labeled DNA primer (15 nM) and template (10 nM) were annealed in 1 × GoTaq buffer (Promega, pH 8.5) with 0.1 mMdNTP by heating at 95ºC for 5 min and were slowly cooled to room temperature. Ligands were added at various concentrations (ranging from 0.1 to 50 μM) and incubated at room temperature for 30 min. Taq DNA polymerase (2.5 U) was added and the mixtures were incubated at 55ºC for 20 min. Some reactions contained 1 µM BRACO19 (a known G-quadruplex-binding compound) as a positive control for quadruplex formation. Data are reported as the average of triplicate experiments. The polymerase extension reactions were stopped by adding 2 × stop buffer (10 mMEDTA, 10 mMNaOH, 0.1% xylene cyanole, and 0.1% bromophenol blue in formamide solution). Samples were heated at 95ºC for 5 min and were loaded onto a 20 × 20 cm 10% denaturing polyacrylamide gel. The gel was run at 800 V for 1 h. After drying the gel and exposing it to a phosphorimager screen overnight, polymerase activity was imaged using a phosphorimager and quantified with Image Quant.
4.6. Circular Dichroism Spectroscopy
CD spectra were recorded on a P-star 180 spectropolarimeter using a quartz cell of 1-mm optical path length and scanned at 25ºC using a wavelength of 220-320nm, a measuring step of 0.5 nm, and a band width of 2.0 nm. The time per point was set to 0.2s and the sample period to 25.5 μs. Before use, the human telomeric oligonucleotide d[G3 (T2AG3)3] was desalted using G-25 Microspin columns following the manufacturer’s instructions (GE Healthcare). The DNA was dissolved in TE (10 mMTris–HCl, pH 7.5, and 1 mMEDTA) at a final concentration of 15 µM in a final volume of 400 µl. The TE buffer included 10 mMLiCl to prevent precipitation of the DNA from the solution. DNA samples were prepared by heating at 95ºC for 5 min and cooling to room temperature. DNA samples were titrated with 0.5 mol equivalents of test compound. After each addition of ligand, the reactions were allowed to equilibrate for at least 15 min to collect the CD spectra. Controls for G-quadruplex formation included 50 mM solutions of Na+ or K+. The compound/DNA ratio varied as follows: 0:1, 0.5:1, 1:1, 1.5:1, 2:1, 2.5:1, 3:1, and 3.5:1.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflicts of interest.
ACKNOWLEDGEMENTS
This research was supported grants from the University Cancer Research Fundof The University of North Carolina (Jarstfer). M Sassano was supported in part by fellowships from Amgen and GlaxoSmithKline. The authors wish to thank the Lingner lab for supplying plasmids pVan107 and pBS-U1-hTR.

