All published articles of this journal are available on ScienceDirect.

ChemVassa: A New Method for Identifying Small Molecule Hits in Drug Discovery
Authors Info & Affiliations
Abstract
ChemVassa, a new chemical structure search technology, was developed to allow rapid in silico screening of compounds for hit and hit-to-lead identification in drug development. It functions by using a novel type of molecular descriptor that examines, in part, the structure of the small molecule undergoing analysis, yielding its “information signature.” This descriptor takes into account the atoms, bonds, and their positions in 3-dimensional space.
For the present study, a database of ChemVassa molecular descriptors was generated for nearly 16 million compounds (from the ZINC database and other compound sources), then an algorithm was developed that allows rapid similarity searching of the database using a query molecular descriptor (e.g., the signature of atorvastatin, below). A scoring metric then allowed ranking of the search results.
We used these tools to search a subset of drug-like molecules using the signature of a commercially successful statin, atorvastatin (Lipitor™). The search identified ten novel compounds, two of which have been demonstrated to interact with HMG-CoA reductase, the macromolecular target of atorvastatin. In particular, one compound discussed in the results section tested successfully with an IC50 of less than 100uM and a completely novel structure relative to known inhibitors. Interactions were validated using computational molecular docking and an Hmg-CoA reductase activity assay. The rapidity and low cost of the methodology, and the novel structure of the interactors, suggests this is a highly favorable new method for hit generation.
INTRODUCTION:
The advent of target-based drug discovery, assisted by advances in x-ray crystallography and NMR, has left drug development companies suffering both from too much and too little information: too much information in the sense that the early push for target discovery has left pharma’s R&D drowning in potentially relevant targets and drug candidates; too little information in the sense that even cutting-edge in silico methods of compound screening failed to produce target-to-compound relational and trending data that could assist in the selection of promising future candidates. At present, the process of taking a candidate compound from a hit to a lead takes approximately two years and costs tens of millions of dollars [1-6]. Early pharmaceutical R&D is in need of new ways to optimize this phase of development using mathematically and biologically sound methods for identifying, ranking, and validating candidate compounds and their relationships to promising biological targets. Vassa Informatics has addressed this need with the development of ChemVassa, an algorithm that allows for rapid in silico screening of compounds for hit and hit-to-lead identification in drug development.
ISSUES AFFECTING DRUG DISCOVERY
The drug discovery process has undergone dramatic change during the last 50 years. Despite many advances, however, only five of 40,000 compounds tested in animals reach human testing. More importantly, only one in five that reach clinical trials is approved. Moving these compounds through the early stages of development represents an enormous investment of time, as well as financial and human resources [4, 6, 7].
Since the 1990’s, advances in synthetic and combinatorial chemistry and laboratory automation have allowed for high-throughput screening of compounds, affording researchers a high probability of uncovering novel molecules. The limitation in this model, however, is the identification of novel targets for therapeutic intervention. Successfully marketed pharmaceuticals produced using this method are primarily “me too drugs”. An excellent example is the statins, which lower cholesterol by targeting the enzyme HMG-CoA Reductase [8]. Today, there are 10 marketed statins, all of which share structural similarity in the HMG moiety. In other words, under this model, successful drug discovery primarily relies on identifying compounds directed against known targets, not on identifying new targets for treating human disease. Further, in spite of increased screening capacity, the rate of newly registered compounds in clinical trials has not kept pace [7, 9].
In parallel to advances in physical screening capability, computational approaches have been developed for identifying potential drug candidates. Screening can be performed in silico using software to “dock” a ligand into the active site of a protein structure. Docking methods attempt to identify the optimal binding position, orientation, and molecular interactions between a ligand and a target macromolecule. Given sufficient computational resources, these “virtual screens” can be performed using very large compound structure libraries, and yield a ligand discovery (hit rate) of two to three orders of magnitude greater than that of empirical screening. Computational approaches also permit the development of focused libraries. In these cases, molecular descriptors are used to transform chemical structures into a standard format that can be used for comparative searching based on a correspondence of the query structure to structures in the database. Such searches often yield molecules with biological activities similar to the query molecule. A powerful extension of the method allows researchers to limit their search results only to those molecules that contain a specific substructure (e.g., a binding pocket or active site). Molecular fingerprints extend this concept by encoding molecular structure in a series of binary digits (bits) that represent the presence or absence of particular substructures in the molecule. Clustering of structures can be used to identify both similarity and dissimilarity (diversity) within a chemical library [10, 11]. The incorporation of other variables or metrics, such as the Lipinski Rule of Five [12, 13], have been used to filter databases and identify compounds with drug-like properties, thus decreasing the rate of compound attrition after initial screening.
While these methods can be used to rapidly develop focused libraries for either virtual or empirical screening, they all suffer from a common problem, in that they all rely on structural similarity to make their evaluations. If searches are based on an existing marketed compound, screening of the resultant library will yield little novelty and potentially introduce issues of intellectual property infringement. Freedom to operate is a critical factor in pharmaceutical development, without which a potential drug cannot achieve marketability.
The identification of novel compounds directed against therapeutic targets of interest remains a major focus for pharmaceutical and biotechnology companies. Although pharmaceutical budgets have increased 20-fold over the past two decades, the rate at which new chemical entities have achieved FDA approval has been on a steady decline (see Fig. 1 below). Many new drugs were introduced in the 1990s to treat previously untreated or undertreated conditions, but the pace of introduction has declined since 2000—in most years, back to levels not seen since the 1980s. The introduction of priority drugs—those that, according to the Food and Drug Administration (FDA), provide a “significant therapeutic or public health advance”—has also slowed, from an average of more than 13 a year in the 1990s to about 10 a year in the 2000s.
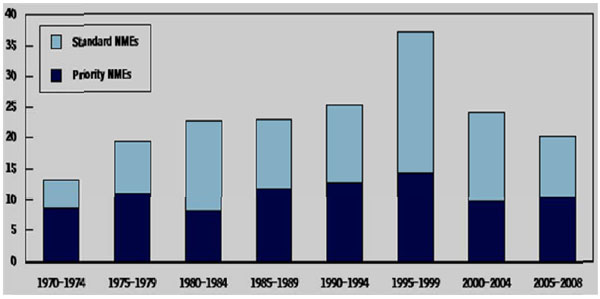
Average Annual Approvals of New Drugs by the Food and Drug Administration, 1970 to 2008. The data, for new molecular entities (NMEs) only, exclude extensions and new approved uses of existing drugs. NMEs are drugs based on a molecule not used before in any pharmaceutical product. Priority drugs are those that, according to the Food and Drug Administration, provide a “significant therapeutic or public health advance.” (Source: Congressional Budget Office based on data from the FDA.)
Between 2004 and 2008, $40 billion in pharmaceutical industry revenues were at risk through patent expiration on just 19 products in the United States alone. Worldwide, a much more dramatic $72 billion stands to be lost. Those revenues will not be replaced primarily by new blockbusters; more likely, in the near future, pharma will be looking at products in the $500 million to $600 million range. If so, pharma will need 80 new molecular entities (NMEs) in the next four years to replace lost US revenue, and 144 to make up for lost revenue worldwide [1, 2, 6].
Two major needs therefore exist within the industry. First is the need to generate new chemical entities to fill the discovery pipeline and address sliding productivity and potential loss of revenue. Second is the need to to shorten development timelines.
ChemVassa addresses these needs through the use of proprietary algorithms that can search chemical databases and identify compounds overlooked by existing methods. The technology evaluates similarity utilizing a unique method for the identification of novel compounds that fingerprints the atom and its position in the molecule, producing its “signature.” This approach affects both attrition rate and the timelines associated with the preclinical drug discovery process. Further, ChemVassa searches can be scaled to any size compound database, inexpensively and efficiently, bringing ultra-high throughput compound screening to both large pharmaceuticals and smaller biotechnology companies, enhancing both throughput and quality for small molecule drug discovery.
NOVEL SCREENING METHODOLOGY
The formulae developed for ChemVassa to footprint small molecule structure content perform two crucial tasks: (1) quantifying a channel’s information potential (channel capacity: the amount of information that a channel is capable of transmitting); and (2) determining the amount of information contained in a signal at the beginning or end of its transmission.
As applied in ChemVassa, the critical metric used to generate a profile of a target molecule considers the structure itself as the information channel, producing a novel metric for evaluating similarities and differences among compounds. Therefore, the location and composition of an atom in a molecule can be considered as contributing to the expansion of a structure (a positive value) or the compaction of a structure (a negative value). With the aid of structural information, ChemVassa is able to create an abstraction of a molecule that shows the contribution of each atom in the molecule to the ability of the molecule to be compressed and represented as a linear string. A low or negative value means that a region or atom is highly compressible; a high value means the region or atom is hard to compress. Our research strongly suggests that rapid fluctuations between high and low values generally characterize functional surfaces, or other notable features of proteins and small molecules. In practice, this approach yields standard structure-based search methods and other structural fingerprinting algorithms [14].
MATERIALS AND METHODOLOGY
The current study, undertaken as a validation of ChemVassa, investigated the technology’s ability to identify novel molecules that were informationally related to a known drug, but still relevant as hit-to-lead compounds. It consisted of three phases: database searches using the information signature of an existing drug to produce a list of hits, molecular docking to verify binding in the top database hits, and finally, bench assays to determine reactivity in the best modeled hits. The query molecular descriptor, or “signature,” used was for atorvastatin (Lipitor™).
CHEMVASSA ALGORITHMS
The ChemVaSSA algorithm works by examining the overall structural and physicochemical similarity of a particular molecule. It does this from a two-fold perspective. First, it utilizes spatial information to locate an atom within a molecule. The spatial information is taken from the source PDB file (or converted SMILES file, e.g.). Each atom is then converted into a chemical lexicon value, which can in practice takes into account the valence shell content, atomic number, and reactivity of the atom. The location of each atom is compared and the reactivity measure is calculated; the distance average and the reactivity comparator gives the “G-score.” G-scores for the molecule are processed as follows: the average for connected molecules is summed and averaged. The result is the “M score.” The M score can be calculated as the average across all molecules (M1) or the average across the carbon chain (M2). In practice, the M1 score is calculated and can be used as a shorthand for referring to the molecules, and the M2 score is currently not calculated for common usage of the program.
G therefore represents whether a particular atom is enhancing or detracting from the information content of the entire molecule. M1 is the average information content attainable across the entire molecule, while M2 represents, loosely, the information content if only the carbon backbone is considered (which may be desirable for peptide-specific studies).
The similarity search algorithm operates by concatenating a model’s G scores into a single bit field and then running bitwise comparisons between different models and subsections of models. The number of bitwise differences between the comparator and the candidate model is used to assess similarity. A logarithmic scale is used to give greater weight to bit differences in the higher-order positions of the bit field.
The ChemVassa database contains a listing of all structure files and models loaded into the system. As each file is loaded, the G-score is computed and stored in the database. Once the file has been loaded, and all G-scores computed, the list of G-scores is extracted from the database. Scores are rounded to the nearest thousandth (which is the maximum resolution used in the PDB file) and scaled to convert the G-scores from decimal to integer notation so as to avoid having to operate on the bitwise IEEE floating-point representation of the scores.
Once G-scores are converted to 32 bit integers, they are concatenated into a single linear bit field and stored in the ChemVassa database for easy recall. These are the signatures, or molecular descriptors, that ChemVassa relies upon to perform its analyses.
DATABASE SEARCH USING MOLECULAR DESCRIPTOR FOR EXISTING COMPOUND (ATORVASTATIN)
We utilized the ChemVassa algorithms to search a database of 5 million compounds (the entire available set of small molecules from RCSB, along with the “goldilocks” and a portion of the “drug-like” sets from the ZINC library [15]—a subset of our 16M compound library). Using the information signature of atorvastatin,we identified ten high-quality hits for further investigation.
Analysis of these hits classified them into three categories based on their potential binding characteristics: (1) known binders, such as other statins, which would be found by existing methods, and serve to validate the functionality of the algorithm (~20%) (all statins included in the screening library were identified, but not all current statins were included in the library screened); (2) false positives, which consisted of complete non-binders, and cases where a portion of the molecule would likely bind except could not due to steric hindrance within the active site of the macromolecule (~40%); and, (3) novel results, which were not structurally similar to atorvastatin, but that appeared to be capable of binding HmG-CoA reductase in a manner similar to atorvastatin based on molecular docking studies (~40%).
The novel hits did not appear to be referenced in the relevant literature. Only one compound had been identified as having potential relevance in the biological pathways involved in cholesterol metabolism. The hits, therefore, continued to molecular docking and eventual bench testing to further validate the ChemVassa approach. None of the compounds identified as novel interactors would have been identified by standard small molecule fingerprinting methodologies, such as Tanimoto fingerprinting, or by standard structural similarity searches [16].
MOLECULAR DOCKING VALIDATION
Docking was performed using the Autodock automated docking software. Autodock requires ligands to be in pdbqt format. The prepare_ligand4.py script from ADT was used to add hydrogens, merge non-polar hydrogens, assign partial charges, and define rotatable bonds for ligands [17]. When required, OpenBabel was used to convert file formats. Glide achieved similar results [18].
The macromolecules were prepared from the PDB file using Autodock Tools (ADT). ADT was used to prepare the receptor coordinate files for AutoGrid and AutoDock. Grid dimensions were based on the identified regions of intermolecular interaction of the ligand within the receptor. Therefore, a series of trial dockings was performed to optimize parameters for docking. The benchmark for optimization was a docking pose that recapitulated binding energy and intermolecular contacts found in the crystallographic structure.
After completion of a virtual screen, the docking log file (.dlg) was parsed using a Python script to extract the calculated free energy of binding. These results were used to rank and select specific binding poses from each ligand for further analysis. Molecular interactions such as bond distances and hydrophobic interactions were calculated and displayed using Pymol, and were compared against the original ligand-receptor data to determine binding quality. Results were then entered in the database, along with the docking parameters and associated metadata. An example result of a promising early hit still being pursued at the bench appears in Fig 2, below.

Analysis of binding of CVSlead12 to the Hmg-CoA reductase site. Panel A shows the molecular interactions o CVSlead12 with the HMG-CoA reductase binding site. Panel B shows the molecular interactions of atorvastatin to the Hmg-CoA reductase site. Panel C shows a comparison table of interactions conserved between A and B. An asterisk indicates a critical bond found in the atorvastatin-HMG-CoA reductase interactions.
The best results from this phase proceeded to the next stage for biological screening.
HMG-CoA REDUCTASE ACTIVITY ASSAYS
For purposes of the assays, 1 ml reaction contained: 100 mM KPi (pH 6.8), 400 mM KCl, 4% DMSO (v/v), 200 uM NADPH, (prepared fresh daily in 100 mM KPi pH 6.8), 0.1 mg/ml BSA, 5 mM DTT, 0.52 to 1.04 ug hHMGCR soluble construct (Sigma H7039) , and 8 uM (R,S) HMG-CoA. DMSO was excluded from KmHMG-CoA assays and IC50 assays of mevinolin, CVSLead1, and CVSLead2.
Cuvettes containing KPi, KCl, BSA, and DTT, were incubated at 37 °C in the spectrophotometer for 10 minutes. NADPH, DMSO, and inhibitor (as needed) were added to the cuvette and absorbance at 340 nm was recorded and allowed to stabilize (~ 2 min). HMGCR was added and allowed to incubate for 2 minutes while a background was recorded.
All reactions were initiated by the addition of HMG-CoA. It is reported that erratic results (non-linear kinetics) are produced when HMG-CoA is used to initiate reactions where a competitive inhibitor of HMG-CoA is present and NADPH should therefore be used to initiate the reaction [19-21]. Reproducible results were not obtained when NADPH was used to initiate the reactions (with or without inhibitor) under the conditions described above. Data were fit by non-linear regression analysis using GraphPad Prizm 4.
Cholic acid and deoxycholic acid were dissolved in 100 mM KPi (pH 6.8). Stock solutions of drug-like compounds (10 mg/ml) were prepared in 100% DMSO. Mevinolin was used as a positive control.
RESULTS
We have completed testing of eight compounds received from Sigma-Aldrich, Ambinter, and Enamine-Real as described in the methodology section. These compounds were initially identified during a screen for small molecules that may have function similar to atorvastatin and other statins, via a screen utilizing the “signature” of atorvastatin in the Chemvassa database. All modeled compounds underwent bench testing for further validation utilizing an HMG-CoA reductase assay screen, available from Sigma-Aldrich, and implemented under the guidance of Dr. Wyckoff at UMKC. Controls were utilized to ensure that we could duplicate the IC50s reported for commercially available statins.
The compounds tested were denoted CVSlead1-8. The results were as follows:
We have verified experimentally that two of eight compounds identified in our survey of the database were, indeed, capable of inhibiting the HMG-CoA reductase enzyme. Further, one hit showed an IC50 of less than 100uM and has favorable properties for further drug development. Example IC50 curves appear in Fig. 3, below. Maximal Hill Slopes were all under 1.5 (using an unconstrained fit) and optimal fit Hill Slopes were well under 1 (using a sigmoidal dose-response model), therefore aggregation is not suspected to play a role in the current results Table 1.
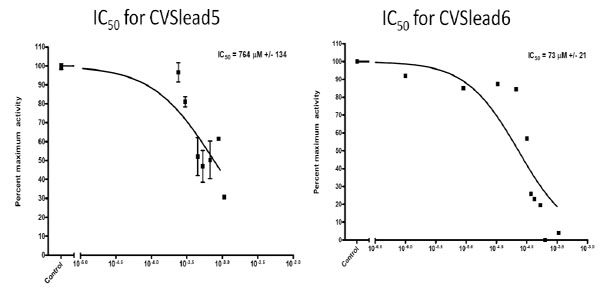
C50 Curves for CVSlead5 and CVSlead6.
Compound Testing Results
| Compound Designation | Inhibits HMG-CoA? | Est. IC50 | Approx. Molecular Weight | H-Donors |
|---|---|---|---|---|
| CVSlead1 | No* | -- | 450 | 3 |
| CVSlead2 | No | -- | -- | -- |
| CVSlead3 | No | -- | -- | -- |
| CVSlead4 | Insoluable under assay conditions | |||
| CVSlead5 | Yes | 510-764uM | 280 | 3 |
| CVSlead6 | Yes | 73uM | 300 | 0 |
| CVSlead7 | No | -- | -- | -- |
| CVSlead8 | Similar to CVSlead4, and presently insoluable—trials continuing | |||
* Solubility may be a factor for thid compound under assay conditions.
The present study has therefore yielded a highly favorable hit ratio, especially given the rapidity of the screening process (approximately 120 days); a majority of the time was spent actually obtaining the compounds for testing, rather than in the screening or testing phase. While CVSLead1 appeared to inhibit HMG CoA reductase in some assays, it was not stable under assay conditions and did not appear to have properties that would be useful for further drug development. We conclude therefore that if we had initially prescreened the database using precalculated physicochemical properties, our hit success ratio would have increased, as compounds with poor solubility (e.g. CVSleads 4 and 8) would have been excluded.
We believe these results validate our screening technique and the Chemvassa methodology,and further, indicates the utility of this method for hit and hit-to-lead screening in drug development.
DISCUSSION AND CONCLUSION
Over the course of the current study, we developed a new methodology for screening large amounts of small-molecule data for determining hits in drug discovery. This methodology, when applied using intelligent filtering and screening methodologies, has produced a high hit-to-lead ratio for the determination of new structures that may be developed to inhibit a well-studied target, HmG-CoA reductase. The identified structures would not have been identified by standard structure search or fingerprinting methods being utilized to date. The relatively low cost per modeled and assayed compound, in addition to the rapid timeframe and novelty of results, are exceptionally favorable and speak to the general applicability of this emerging technology.
ABBREVIATIONS
CONFLICT OF INTEREST
The authors confirm that this article content has no conflicts of interest.
ACKNOWLEDGEMENTS
The authors wish to thank the members of the Miziorko lab for help in obtaining screening results, Christine Malcom at Roosevelt University for reviewing early drafts of the manuscript, and members of the Amazon Web Services team for help in setting up screening libraries in their cloud computing resource. This work was supported in part by NIH grant NIGMS089922 to Dr. Wyckoff.

