All published articles of this journal are available on ScienceDirect.

Recent Progress in the Use of Glucagon and Glucagon Receptor Antago-nists in the Treatment of Diabetes Mellitus
Authors Info & Affiliations
Abstract
Glucagon is an important pancreatic hormone, released into blood circulation by alpha cells of the islet of Langerhans. Glucagon induces gluconeogenesis and glycogenolysis in hepatocytes, leading to an increase in hepatic glucose production and subsequently hyperglycemia in susceptible individuals. Hyperglucagonemia is a constant feature in patients with T2DM. A number of bioactive agents that can block glucagon receptor have been identified. These glucagon receptor antagonists can reduce the hyperglycemia associated with exogenous glucagon administration in normal as well as diabetic subjects. Glucagon receptor antagonists include isoserine and beta-alanine derivatives, bicyclic 19-residue peptide BI-32169, Des-His1-[Glu9] glucagon amide and related compounds, 5-hydroxyalkyl-4-phenylpyridines, N-[3-cano-6- (1,1 dimethylpropyl)-4,5,6,7-tetrahydro-1-benzothien-2-yl]-2-ethylbutamide, Skyrin and NNC 250926. The absorption, dosage, catabolism, excretion and medicinal chemistry of these agents are the subject of this review. It emphasizes the role of glucagon in glucose homeostasis and how it could be applied as a novel tool for the management of diabetes mellitus by blocking its receptors with either monoclonal antibodies, peptide and non-peptide antagonists or gene knockout techniques.
INTRODUCTION
Glucagon is a hormone made up of 29 amino acids. It is formed from pro-glucagon precursor. Proglucagon is proteolytically processed into various peptide hormones such as glucagon in pancreatic alpha cell, glucagon like peptide-1 (GLP-1), GLP-2, oxyntomodulin and glicentin in intestinal L cells and the central nervous system [1]. Diabetes mellitus (DM) is characterized by chronic increment of fasting blood glucose level associated with marked and sustained postprandial hyperglycemia. At the same time, type 2 diabetes mellitus (T2DM) is associated with a disturbed secretion and function of several pancreatic hormones including insulin and glucagon all of which play an essential role in glucose homeostasis. T2DM is associated, especially in late stage, with a deficiency in insulin secretion and impairment in the ability of insulin to trigger its receptors effectively and help in the uptake of glucose into specific cells. The deficiency of insulin secretion and insulin resistance are correlated with reduction of glucose uptake in certain cells such as those of skeletal muscle. This condition is associated with hyperproliferation of pancreatic, glucagon secreting alpha cells resulting in hyperglucagonemia [2-4]. Several modified molecules of glucagon have been shown to inhibit the function of glucagon receptors, thereby preventing the hyperglucagonemia-induced hyperglycemia seen in DM [3, 5]. Insulin itself is a glucagon antagonist, inhibiting glucagon secretion via the activation of GABA receptor and its translocation to the cell membrane to prevent glucagon release [6]. The low insulin level observed in diabetic patients will result in high level of glucagon because of loss of insulin-induced glucagon inhibition.
Glucagon receptor belongs to the B-family of the trans-membrane receptors called G-protein- coupled receptor (GPCR) [7]. The receptors responsible for the function of key bioactive agents such as GLP-1 and GLP-2, and glucose-dependent insulinotropic peptide (GIP) are active members of this group of receptors as well [8].
Glucagon promotes glucose production from the liver, mainly via binding with the G-protein coupled glucagon receptor to stimulate gluconeogenesis and glycogenolysis through the adenylate cyclase that initiates protein kinase A (PKA) activity. Simultaneously, PKA suppresses glycogen synthesis and glycolysis and induces ketogenesis [9]. Glucagon also initiates lipolysis in the liver and adipose tissues [10]. Glucagon receptors are expressed not only in the liver but also in many other tissues such as heart, smooth muscle of the intestine, brain, kidney and adipose tissues [11].
GLUCAGON AND DIABETES
The hormone glucagon plays a major role in the initiation and elevation of hyperglycemia in DM. Insulin and glucagon antagonize each other in the regulation of glucose level. In DM the blood plasma ratio of glucagon versus insulin levels in the blood plasma is higher than that observed in healthy controls. Although previous studies have reported that diabetic patients have significant reduction in the number of glucagon receptors, however, the effect of this peptide hormone at its receptor sites and the ability to activate cAMP remains largely intact [12]. This is probably due to the fact that the activation of 20% of glucagon receptors is enough to significantly stimulate adenylate cyclase [13]. Type 1 and 2 DM are associated with hyperglycemia, due in part to lack of suppression of glucagon release because of insulin loss [9, 14].
GLUCAGON RECEPTOR
The binding of glucagon to its GPCR, located on the plasma membrane results in conformational alterations in the nature of GPCR. The Gαs and Gαq subunits of GPCR play an important role in glucagon-induced cell signaling in selected cells. Activation of Gαs receptor induces large and significant increases in adenyl cyclase, and consequently in the cytoplasmic levels of cAMP and PKA. Moreover, stimulation of Gαq induces large increases in the level of phospholipase C, synthesis of inositol 1,4,5-triphosphate, followed by the release of calcium ion intracellularly, leading to glucose release from hepatocytes through initiation of glycogenolysis, gluconeogenesis and glycolysis and suppression of glycogenesis [15]. Abolition of glucagon receptors (Gcgr−/−) leads to reduction in plasma glucose levels [16].
GLUCAGON RECEPTORS IN THE TREATMENT OF DIABETES
The pathogenic symptoms of DM, such as hyperglycemia and ketoacidosis are mainly due to the excessive circulating glucagon levels, while deficient insulin level or insulin resistance leads to decreased uptake of glucose. This physiological role of glucagon encourages many investigators to focus on putative glucagon antagonists to restore normoglycemia in patients suffering from DM. In view of this, glucagon receptor antagonists or molecules that could suppress the expression of glucagon receptors have attracted the attention of investigators working on the treatment of DM [17].
GLUCAGON RECEPTOR GENE KNOCKOUT AS A TOOL FOR VALIDATING THIS RECEPTOR AS A POTENTIAL DRUG TARGET
The fact that glucagonemia is not a good sign in DM has prompted investigators to try and block glucagon receptors on hepatic cells. This idea has stimulated scientists to construct mice lacking glucagon receptors Gcgr−/−. The inability of Gcgr−/− mice to develop hyperglycemia have supported the role of glucagon and its receptors in the pathogenesis of diabetes-induced hyperglycemia. Glucagon receptor gene knockout Gcgr−/− mice under normal diet showed a decreased blood glucose level. This Gcgr−/− mice also resist streptozotocin-induced hyperlycemia and show less beta cell damage [18]. In other studies, blocking of glucagon action (absence of glucagon receptor) in Gcgr−/− streptozotocin-induced diabetic mice leads to enhancement of metabolic state and this finding indicates that glucagon receptor gene deletion in type 1 diabetic mice restores its normal metabolism [19]. Fasted Gcgr−/− mice also showed a decrease in blood glucose level but not an overt hypoglycemia, because increase in blood glucose is not regulated by glucagon alone but also by several other hormones including catecholamines, corticosteroids and growth hormone [20]. However, fasted Gcgr−/− mice showed considerable deficiency in lipid production, secretion, and oxidation [21]. This result indicates that a minimal essential level of glucagon receptor expression is needed to maintain normal lipid metabolism in the liver [21]. In contrast, other studies have shown that the level of lipids is normal in the presence of lower glucagon receptor level [22].
Gcgr−/− mice were characterized by enhanced glucose tolerance and associated with increased GLP-1 plasma concentrations. This elevation of GLP-1 is mainly produced by alpha cells [16]. The improvement of glucose homeostasis in these Gcgr−/− mice resulted mainly from either suppression of glucagon signaling or increased secretion of GLP-1 [23].
Suppression of liver Gcgr expression was seen after treatment of db/db mice with antisense oligonucleotide. Reduction of liver Gcgr expression was associated with decreased blood glucose concentration, plasma free fatty acids and triglycerides with undetected hypoglycemia [24].
In addition, Gcgr antisense oligonucleotide administration to ob/ob and db/db mice and Zucker diabetic fatty rats showed a reduction of both blood glucose levels and liver triglycerides. Also, treatment of rodents via Gcgr antisense oligonucleotide leads to increased blood concentration of glucagon and GLP-1 with hyperproliferation of alpha cells [25]. This investigation in rodents revealed that the suppression of Gcgr expression and glucagon effect lead to a reduction of liver glucose output with enhanced glucose tolerance [26].
Gcgr−/− mice of either streptozotocin-provoked or high-fat diet treated has been investigated. The results of glucagon receptor deletion mice compared to normal control mice had showed that Gcgr−/− mice were characterized by reduced body weight, appetite and gastric motility with decreased blood glucose concentrations and enhanced glucose homeostasis. In addition, Gcgr−/− mice showed decreased hyperleptinaemia and liver steatosis induced by high-fat diet and less susceptible hyperglycemia in streptozotocin-induced beta cell damage [18].
Prominent alpha cell hyperplasia and elevated blood glucagon levels were seen in Gcgr−/− mice as a compensatory effect due to deletion of Gcgr. Moreover, a reduction in free fatty acid and β-hydroxybutyrate concentrations were seen in Gcgr−/− mice [19].
ANTIBODY NEUTRALIZATION OF GLUCAGON
Many investigators have reported that suppression of glucagon action causes considerable improvement in elevated blood glucose concentrations in animal models of DM. Immunoneutralization of glucagon by monoclonal antibody in diabetic rats, mice and rabbits lead to a significant decrease in blood glucose levels. Moreover, there was a significant reduction in serum levels of glycosylated hemoglobin A1c and serum triglyceride in ob/ob mice treated with glucagon antibodies [18-21]. In addition, glucagon antibodies significantly decrease the output of glucose from the liver, which in turn reduces the elevated blood glucose levels in diabetic and normal rabbits [20]. Other studies reported that glucagon immunoneutralization leads to a marked decrease in glucagon levels and improve glucose tolerance in experimental animals [18, 19]. It is clear from these studies that immunoneutralization of glucagon may be a promising tool for the treatment of DM (Table 1).
The use of immunoneutralization of glucagon receptors by monoclonal antibody.
| Dosage | 1 - 30 mg/kg (monoclonal antibody) | References |
|---|---|---|
| Mode of delivery | Subcutaneous (s.c.) | [18-21] |
| Efficacy | Single s.c. injection of 3 mg/kg body weight maintained normoglycemia for 8 days. 30 mg/kg body weight achieved a longer duration of normoglycemia. No hypoglycemia was recorded. Monoclonal antibody against glucagon receptors was ineffective in the face of severe hyperglycemia without the addition of insulin. |
Peptide antagonists of glucagon receptors.
| Dosage | Mode of Delivery | Efficacy | References | |
|---|---|---|---|---|
| Bicyclic 19-residue peptide BI-32169 | 320-440 nM | Subcutaneous (s.c.) or intravenous (i.v.) | Investigations still in the experimental phase. | [42, 43] |
| Des-His(1)-[Glu(9)]-glucagon amide | 10 µg | Intravenously (i.v.) | Single dose blocks up to 40-80% of endo- as well as exogenous glucagon, including free as well as hepatocyte-bound. | [39,40, 43-45] |
The advantages and disadvantages of glucagon neutralization and glucagon receptor antagonists.
| Advantages | Disadvantages | References | |
|---|---|---|---|
| Neutralizing antibodies against glucagon | Reduced hyperglycemia; lowers triglyceride concentration | Weight gain | [22] |
| Glucagon receptor antagonism | Decreased blood glucose concentrations | Abnormal lipid production | [16, 21] |
The effect some glucagon receptor antagonists.
| Drug | Compound | Efficacy | Dosage | Adverse effect | Clinical phase | Ref |
|---|---|---|---|---|---|---|
| Spiro-urea | Compound 15 | (a)Reduced glucose excursion after glucagon challenge ; (b) blocks glucagon effect | 10 and 30 mg/kg (i.m.) | thirst, dry mouth, nausea, polyuria | Pre-clinical | [53, 67] |
| Benzimidazole | Compound 36 | Reduced glucose levels in diabetes; suppressed glucagon-induced glucose increase in animals | 10 and 3 mg/kg in rhesus monkeys |
Reduced human liver microsome stability | Pre-clinical | [56, 68] |
| Skyrin | Fungal bisanthro-quinone | Antagonized functional glucagon (uncouples glucagon receptor from adenylate cyclase in hepatocyte plasmalemma | 30 micromol/l inhibited glucagon-stimulated cAMP production and glucose output | Suppressed Na+, K+-activated ATPase; Inhibits microsomal activity in rat brain | Not in clinical trial | [51, 69] |
PEPTIDE ANTAGONISTS OF GLUCAGON RECEPTORS
Glucagon antagonists inhibit both glucagon receptors binding and the action of glucagon at the receptor binding site. Some glucagon antagonists may inhibit glucagon action via competitive or noncompetitive pathways [31, 32]. Glucagon receptors show species specificity as some of them are more effective on human glucagon receptor than those of rodents [33]. According to many reports, experimental animal expressing receptors similar to those of human will be a potent tool to demonstrate the in vivo estimation of the effectiveness of glucagon receptor antagonists in the treatment of human DM [34].
Many recent studies were directed towards the discovery of new ways of suppressing glucagon action using glucagon receptor antagonists with a strong binding activity towards glucagon receptors than the native glucagon [35-37]. The administration of glucagon receptor antagonists leads to a reduction in blood glucose levels in normal and diabetic rodent models [38-40]. A number of glucagon antagonists have recently been reported. Many studies were focused on the discovery of glucagon peptide derivatives of potent glucagon receptor antagonist through the modification of different amino acids moiety in native glucagon hormone. Many glucagon derivatives studied include His1, Phe6, Ser8, Asp9, Tyr10, Ser11, Lys12, Tyr13, Asp15, Ser16, Arg17,18, Asp21 and Trp25 [41] and bicyclic 19-residue peptide BI-32169, Des-His(1)-[Glu(9)]-glucagon amide. This naturally occurring peptide was isolated from Streptomyces sp [42]. Administration of this bicyclic 19-residue peptide BI-32169 showed a strong reduction in human glucagon receptor activity in a cell-based experiment [43]. Bicyclic 19-residue peptide BI-32169 novel peptide is considered to belong to the lasso group. The potential advantage of this compound is the fact that it is a naturally occurring substance (Table 2).
Many investigators have tried to design a glucagon receptor antagonist by modifying the sequence of its amino acid. The des-His(1)-[Glu(9)]-glucagon amide is an outcome of this endeavor. The glucagon receptor antagonist des-His(1)-[Glu(9)]-glucagon amide was reported to totally abolish the activity of glucagon receptor and leads to a reduction in hyperglycemia in normal rabbits and in streptozotocin-induced diabetic rats when administered intravenously [43, 44]. Des-His-glucagon, a peptidyl glucagon receptor antagonist, binds to about 80% of the mice liver glucagon receptors and prevents the increase in glucagon-induced plasma glucose [39]. Other glucagon receptor antagonist [1-natrinitrophenylhistidine, 12-homoarginine]-glucagon showed a marked reduction (20-35%) of blood glucose levels in streptozotocin-induced diabetic rats when given intravenously [40]. Similar antagonistic effect was reported by des-His, des Phe(6),[Glu(9)]-glucagon-NH2, which also has hypoglycemic effect. 750 µg/Kg body weight induced up to 63% decrease in the level of hyperglycemia, when given intravenously [45] (Table 2).
NON-PEPTIDE GLUCAGON RECEPTOR ANTAGONISTS
Many orally administered doses of small molecules such as ureas, beta-alanine derivatives, alkylidene hydrazides and benzimidazole were reported to be able to block glucagon receptor in both non-diabetic and diabetic dogs, and monkeys [38-40]. Recent studies have shown that beta-alanine urea derivatives can block glucagon from binding to human glucagon receptor when given intragastricaly at a dose of 20-100 mg/kg [46, 47]. Beta alanine, also known as 3-aminopropanoic acid, is a non-essential amino acid that is frequently used by sportsmen to enhance their performance. (+)-3,5 diisopropyl-2-(1-hydroxyethyl)-6-propyl-4'-fluoro-1,1'- biphenyl; C23H31FO) (Bay 27-9955) is a small non-peptide glucagon receptor antagonist, which has been reported to prevent hyperglucagonemia when administered intravenously at a dose of 70-200 mg. However, Bay 27-9955 can also be given orally. It prevents glucagon-induced increase in glucose release from the human liver in a dose-dependent way [48]. See Fig. (1) for the structure of some selected glucagon receptor antagonists.
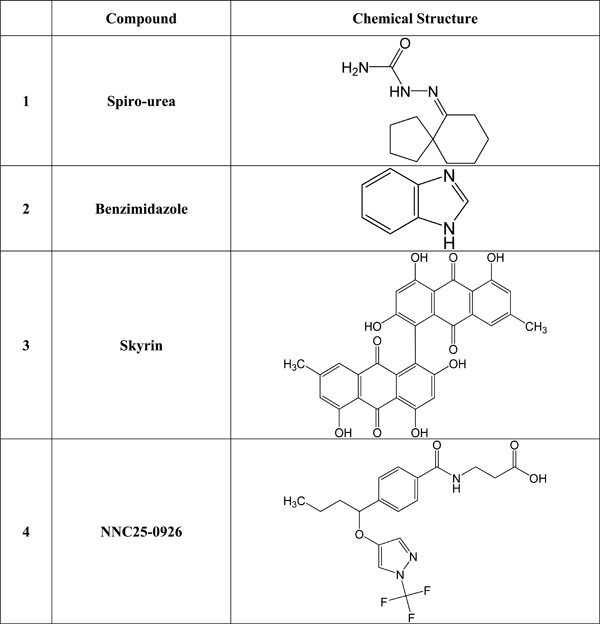
Chemical structure of selected glucagon and/or glucagon receptor antagonists.
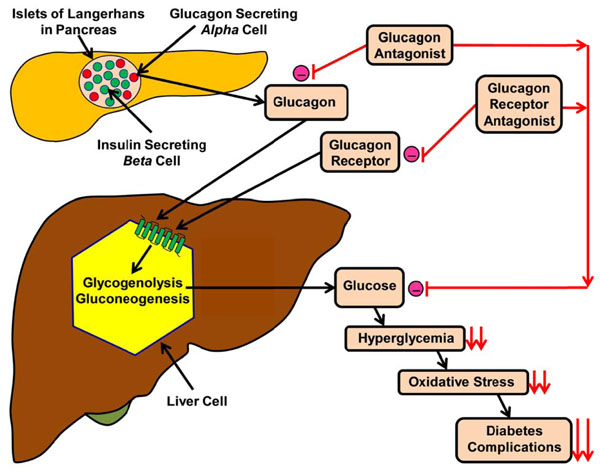
Effects of either glucagon or glucagon receptor antagonist on reduction of diabetic complications. Fig. (2). shows how a schematic diagram of interaction between glucagon, glucagon antagonist, glucagon receptor antagonist and glycogenolysis in the regulation of blood glucose level. (-) = inhibition, = ↓↓reduction.
One of the other non-peptide glucagon receptor antagonists is a 5-hydroxyalkyl-4- phenylpyridines which has about 70-fold more binding capacity to the human glucagon receptor compared to wild glucagon hormone [49]. In addition, compound-1 (Cpd1) is one of the most effective glucagon receptor antagonists that can bind glucagon in human liver cells. Cpd1 also leads to a reduction in glucagon-stimulated glucose increase in mice liver when given intraperitoneally, at a dose of 15 mg/Kg body weight. Cpd1is an effective tool in the reduction of hepatic glucose release and decreasing hyperglycemia in type 2 DM [50].
Skyrin, a fungal product, is a low molecular weight non-peptide glucagon receptor antagonists which does not bind to glucagon receptors but act only as an inhibitor of glucagon-stimulated cAMP activation and glycogenolysis, via uncoupling or disconnect the glucagon receptor from adenylate cyclase activation, thus preventing the glucagon receptor from the production of cAMP that inhibits glucose release from the liver. The effective dose of skyrin is 10-30 mM [51].
In addition to skyrin, other agents including N-[3-cano-6-(1,1-dimethylpropyl)-4,5,6,7-tetrahydro-1-benzothien-2-yl-]-2-ethylbutamide, has been examined. It was shown that N-[3-cano-6-(1,1-dimethylpropyl)-4,5,6,7-tetrahydro-1-benzo-thien-2-yl]-2-ethylbutamide inhibits the attachment and function of the glucagon-receptor complex thereby suppressing the action of glucagon at 15 µg (i.p.) and act as glucagon receptor antagonists [50]. Compounds such as (2R)-N-[4-({4-(1-cyclohexen-1-yl)[(3,5-dichloroanilino) carbonyl] anilino} methyl) benzoyl]-2-hydroxy-balanine, (NNC 25-0926) has been shown to inhibit hepatic production of
glucose when administered at a dose of between 10 and 100 mg/Kg body (intragastrically). See Fig. (1) for the structure of selected glucagon receptor antagonists.
A study of the activity of urea biaryl amide derivative 6i as glucagon receptor antagonist on Sprague-Dawley rats has showed 50% inhibition of glucagon action on glucose liver output under glucagon challenge [52]. Similarly, a spiro-ureas substitution, compound 15, has revealed as inhibitory action as glucagon receptor antagonist on genetically modified mice expressing human glucagon receptor with moderate reduction of hyperglycemia provoked by glucagon [53]. In a related study, a trisubstituted urease derivative, compound 4a, has demonstrated its potent suppression of glucagon receptor action in glucagon-challenged mice expressing human glucagon receptor. This treatment leads to lowering plasma glucose concentrations under high lipid diet [54].
A 1,3,5-pyrazoles substitution, compound 26, has showed a strong glucagon receptor antagonists on rhesus monkey and mice expressing human glucagon receptor. Orally administered compound 26 reduced the hyperglycemia under glucagon effect [55].
Oral effective aminobenzimidazole glucagon receptor antagonist has been established as a glucagon suppressor drug. Aminobenzimidazole showed reducing effect on glucose concentrations in glucagon-induced murine diabetic model, rhesus monkeys, and genetically modified human glucagon receptor mice [56].
A study of the pharmacodynamics effect of orally β-alanine derivatives as glucagon receptor antagonists lead compound 25 and the optimized derivative 57 had revealed that they suppressed the glucagon-challenged hyperglycemia in rats mainly through reduction of glucagon action on liver glucose production [57].
A promising glucagon receptor antagonist compound 15 is derived from β-alanine series by changing the β-alanine motif to isoserine. Compound 15 has confirmed as suppressor glucose output under glucagon-challenge from rat liver cells. In addition, compound 15 reduced hyperglycemia in Sprague-Dawley rats and in ob/ob mouse [46].
Epigenetic changes occur via a variety of mechanisms including, chromosome conformational modification, histone alteration, other DNA-associated proteins and non-coding RNAs [58].
Epigenetic alterations may be caused by methylation of chromosomes. This in turn may either activate or suppress relevant genes [59]. Other ways in which epigenetic variation may occur is by histone post-translation alterations such as acetylation, citrullination, cutting, methylation, phosphorylation and ubiquitination [58]. Alterations in histone structure can change the way that histone binds or interacts with other proteins that modify transcriptional mechanism [60]. Furthermore, non-coding RNAs association with DNA can change chromosomal structure eventually leading to alterations in post-translational gene expression [61].
Chromosomal alterations may contribute to epigenetic variants that are involved in genomic modifications associated with some cancers and others diseases [62]. It has been shown that epigenetic chromosomal methylation influences type 1 diabetes via alteration of insulin and interleukin genes [60]. Epigenetic changes leading a decrease in the gene expression of either Glut4 or Pdx-1 has been observed in T2DM [63-65]. Moreover, it has been shown that chromosomal modification accompanied with histone acetylation can cause the progression of DM and thus, the use of histone deacetylase inhibitors as an epigenetic therapy can suppress the development of DM [66]. Other agents that prevent unwarranted DNA methylation and other molecular factors leading to epigenetic changes will be useful in the management of DM. In fact it has been reported that GLP-1 and GIP, molecules related to glucagon can overturn the epigenetic phenomenon in T2DM [64]. In fact it has been shown that GLP-1 and GIP act via GPCR, the receptor through which glucagon exerts its action [7].
CONCLUSION
Optimal management of diabetic patients using glucagon antagonist must avoid the adverse effects of this drug, such as hypoglycemia, liver stenosis, increases in liver enzymes, and disturbed lipid metabolism. Accordingly, reduction of blood glucagon level by using glucagon receptor antagonists must balance between therapeutic benefits and adverse effects.
This mini-review has shed light on the role of glucagon in the regulation of glucose metabolism and how it could be used as a tool for the treatment of DM either by using antagonists or gene knockout techniques that could neutralize the action of glucagon or blocking the binding of glucagon to its receptors with antibody immunization (Fig. 2 and Table 3) A summary of the efficacy, dosage, adverse effect and the status of some glucagon receptor antagonists is given in Table 4.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
Declared none.
PATIENT’S CONSENT
Declared none.

