All published articles of this journal are available on ScienceDirect.

Design and Synthesis of Novel Arylketo-containing P1-P3 Linked Macro-cyclic BACE-1 Inhibitors
Authors Info & Affiliations
Abstract
A series of arylketo-containing P1-P3 linked macrocyclic BACE-1 inhibitors were designed, synthesized, and compared with compounds with a previously known and extensively studied corresponding P2 isophthalamide moiety with the aim to improve on permeability whilst retaining the enzyme- and cell-based activities. Several inhibitors displayed substantial increases in Caco-2 cell-based permeability compared to earlier synthesized inhibitors and notably also with retained activities, showing that this approach might yield BACE-1 inhibitors with improved properties.
INTRODUCTION
Alzheimer’s disease (AD) is a devastating neurodegenerative disorder and the most common form of dementia among the elderly. It is believed that 24 million people worldwide suffer from the disease [1], and with an aging population the number of patients with AD is expected to rise. To develop disease-modifying drugs is therefore of paramount importance. The pathological findings associated with AD are insoluble extracellular amyloid β-plaques, consisting of Aβ-peptides, and intracellular neurofibrillary tangles found in the brain [2]. The human aspartic protease BACE-1 (β-secretase) initiates the cleavage of the amyloid precursor protein (APP) followed by γ-secretase which generates the Aβ-peptides, where BACE-1 catalyzes the rate-limiting step in this sequence [3]. This, together with the knowledge that BACE-1 knockout mice are viable and fertile, suggests that inhibition of BACE-1 could be a promising therapeutic target for the treatment of AD [4, 5]. However, the development of potent and selective BACE-1 inhibitors able to cross the blood-brain barrier (BBB) and avoid P-glycoprotein (P-gp) efflux shuffling has proven to be challenging [6, 7]. Notably though, in December 2013, Merck announced that they plan to initiate a new phase III study for their BACE-1 inhibitor MK-8931. Also, in September 2014 AstraZeneca and Eli Lilly & Company announced that they will develop and commercialize AZD3293. This proves that it is possible to design and synthesize potent and selective BACE-1 inhibitors [8].
Earlier studies in our laboratory [9] led to the development of a series of macrocyclic BACE-1 inhibitors, e.g. compound 1a (Fig. 1), where both promising enzymatic and cell-based activities were observed. However, as these inhibitors suffered from low permeability, we have in this study focused on improving the cell-based permeability by replacing the terminal amide of the P2 isophthalamide moiety with an acetophenone-type ketone group, thus rendering the molecules less peptidic (Fig. 1). Earlier studies have shown that the amide carbonyl oxygen of the P2 isophthalamide moiety forms a hydrogen bond with Thr232 in BACE-1, whilst the amide nitrogen does not make significant interactions with the enzyme [10, 11]. Moreover, it has been shown that the amide group of the P2 isophthalamide moiety can be replaced by a carbon chain, without significant loss in potency [6, 11], which renders the BACE-1 10s loop in a closed conformation with a hydrogen bond between Ser10 and Thr232, stabilizing the closed conformation. We thus hypothesized that an acetophenone-type carbonyl would provide an advantageous bioisostere to the amide group in this position, resulting in higher permeability through making less overall hydrogen bonds and provide inhibitors with retained good activity compared to their isophthalamide analogues. Also, further attempts to improve the cell-based permeability were performed by replacing the P2 sulfonamide functionality with a less polar methyl group.
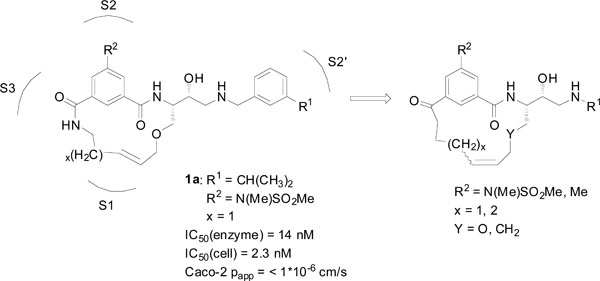
Lead compound 1a together with a general structure of the target macrocycles presented in this paper.
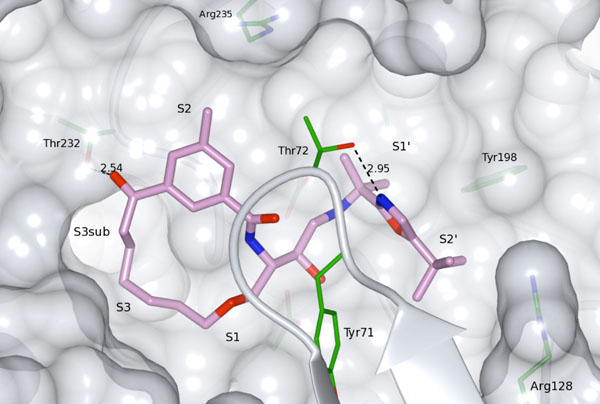
Compound 33 bound to the active site of BACE-1 (PDB id: 4GMI).

Synthesis of building block 8. Reagents and conditions: (i) AcOH (aq.), 60 °C, 3 h, 78%; (ii) TEA, Bu2SnO, p-TsCl, dry DCM, rt, 3.5 h, 74%; (iii) K2CO3, MeOH, 0 °C to rt, 3 h, 84%; (iv) 3-isopropyl-benzylamine, TEA, MeOH, 30 °C, 44 h, 56%; (v) Boc2O, TEA, MeOH, rt, 1 h, 81%; (vi) 1,3-propanedithiol, TEA, Aliquat 336, MeOH, rt, 6 days, 76%.

Synthesis of building blocks 18a-b and 19. Reagents and conditions: (i) pent-4-enylmagnesium bromide or hex-5-enylmagnesium bromide in THF, Et2O (dry), -30 °C to rt, 21 h, 80% and 87%, respectively; (ii) p-nitrobenzoic acid, DIAD, PPh3, THF, rt, overnight, 91%; (iii) NaOMe, MeOH, rt, 1 h 45 min, 68%; (iv) DPPA, PPh3, DIAD, THF (dry), -30 °C to rt, 24 h, 98% and 88%, respectively; (v) 70% AcOH (aq.), 60 °C, 3 h, 74% and quant., respectively; (vi) TEA, Bu2SnO, p-TsCl, DCM, DMF, rt, overnight, 84% and 88%, respectively; (vii) Na2CO3, MeOH, 0 °C to rt, 24 h, 100% and 97%, respectively; (viii) 3-isopropylbenzylamine, TEA, MeOH, 30 °C, 24 h, 61% and 53%, respectively; (ix) Boc2O, TEA, MeOH, rt, 1.5 h, 97%; (x) 1,3-dipropanedithiol, TEA, aliquat 336, MeOH, rt, 5 days, 83%, 81%, and 88%, respectively.
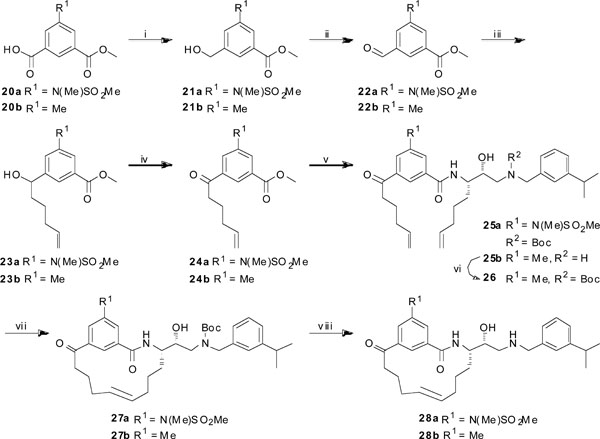
Synthesis of target compounds 28a and 28b. Reagents and conditions: (i) BH3SMe2, THF (dry), Ar, 0 °C to rt, 24 h, 95% and 69%, respectively; (ii) SO3-pyridine, TEA, DMSO (dry), rt, 1.5 h, 84% and 73%, respectively; (iii) pent-4-enylmagnesium bromide in THF, Et2O (dry), 0 °C, 3 h, 65% and 79%, respectively; (iv) Dess-Martin periodinane, DCM, rt, 24 h, 76% and 89%, respectively; (v) (a) LiOH (aq.), dioxane, rt, 3 h; (b) compound 19 or 18a, HATU, DIPEA, DMF, 0 °C to rt, 24 h, 89% and 85% over two steps, respectively; (vi) Boc2O, TEA, MeOH, rt, 3 h, 65%; (vii) Hoveyda-Grubbs 2nd generation, DCM (dry), reflux, 24 h, 21% and 47%, respectively; (viii) Et3SiH, TFA, DCM, rt, 1 h, 91% and 91%, respectively.
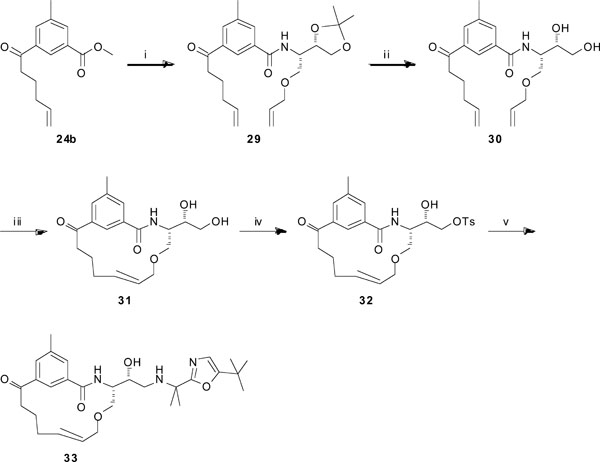
Synthesis of target compound 33. Reagents and conditions: (i) (a) LiOH (aq.), dioxane/H2O, 6 h; (b) (S)-2-(allyloxy)-1-((S)-2,2-dimethyl-1,3-dioxolan-4-yl)ethanamine, HATU, DIPEA, DMF, 4 h, 57% over two steps; (ii) 80% AcOH, 60 °C, 1 h, 95%; (iii) Hoveyda-Grubbs 2nd generation, DCM (dry), reflux, 16 h, 88%; (iv) TEA, Bu2SnO, p-TsCl, DCM, DMF, rt, overnight, 57%; (v) 2-(5-(tert-butyl)oxazol-2-yl)propan-2-amine, DIPEA, EtOH, reflux, 4 days, 5%.
We have also investigated the effect of replacing the oxygen atom in the macrocyclic ring structure, used in some of our earlier inhibitors [9], with a carbon atom and in the P2´ position replacing the 3-isopropylbenzylamine moiety with 2-(5-(tert-butyl)oxazol-2-yl)propan-2-amine, aiming at improving the overall pharmacokinetic properties.
RESULTS AND DISCUSSION
Chemistry
The target compounds 28-36 were prepared according to Schemes 1-4 (see also Table 1). The synthesis of compound 8 is outlined in Scheme 1. Compound 2 was synthesized according to an already published procedure [9]. The acetal in 2 was hydrolyzed with AcOH (aq.) at 60 °C to give the diol 3 in 78% yield. The primary hydroxyl group in 3 was selectively tosylated using p-TsCl, Bu2SnO, and TEA in dry DCM [12] to generate compound 4 (74%). The tosylated compound was then converted into the corresponding epoxide 5 in 84% yield, using K2CO3 in MeOH. The epoxide was thereafter opened with 3-isopropylbenzylamine furnishing compound 6 in 56% yield. For purification reasons, the prepared amine was protected using Boc2O and TEA in MeOH to give compound 7 (81%). Finally, the azide 7 was reduced using 1,3-propanedithiol, TEA, and Aliquat 336 in MeOH to afford the amine 8 in 76% yield.
BACE-1 inhibition data and cell permeability values.
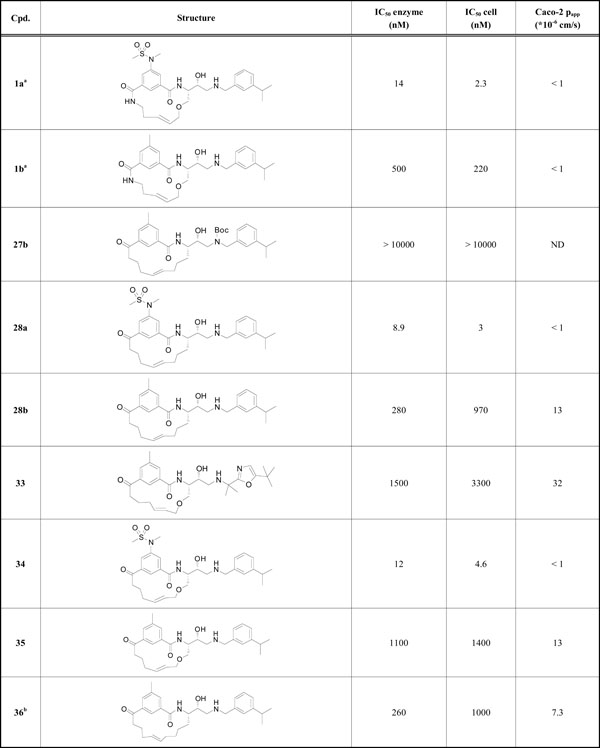 |
a Previously reported by our laboratory
b 16-membered macrocycle
The synthesis of compounds 18a, 18b, and 19 is depicted in Scheme 2. 2,3-O-Isopropylidene-D-glyceraldehyde was synthesized according to a literature procedure [13] and the addition of a freshly prepared Grignard reagent, pent-4-enylmagnesium bromide or hex-5-enylmagnesium bromide, produced the two compounds 9a and 9b in diastereomeric mixtures which were subsequently separated. In the case of 9b we had enough material of the correct diastereomer after the chromatography separation to proceed directly to 12b. (The various stereoisomers were identified by NMR analyses). The hydroxyl group in (S,R)-9a was reacted with PPh3, DIAD, and p-nitrobenzoic acid in THF, furnishing compound 10a in 91% yield. The benzoate ester was cleaved using NaOMe in MeOH to give the inverted alcohol 11a in 68% yield. Mitsunobu conditions, utilizing DPPA [14], generated the azide 12a with inversion of configuration in 98% yield. The acetal in 12a was hydrolyzed in AcOH (aq.) at 60 °C (74%) and the primary alcohol in the formed diol 13a was selectively tosylated, using TEA, Bu2SnO, and p-TsCl in DCM and DMF, to give compound 14a in 84% yield. The epoxide 15a was prepared in 90% yield using Na2CO3 in MeOH, and was then opened with 3-isopropylbenzylamine and TEA in MeOH at 30 °C to give compound 16a (61%). Compound 16b was synthesized, with comparable yields, in the same manner from 12b. Finally, reduction of the azide group in compounds 16a, 16b, and 17 was achieved using 1,3-propanedithiol, TEA, and Aliquat 336 in MeOH, providing the amines 18a and 18b in 83% and 81% yield, respectively. As an alternative synthetic route, for purification reasons, compound 16a was treated with Boc2O and TEA in MeOH to afford the Boc-protected compound 17 in 97% yield. 17 was then reduced as above, yielding 19 (88 %).
The synthesis of target compounds 28a and 28b is outlined in Scheme 3. The compounds were synthesized using two slightly different routes, depending on if the amine (see Scheme 2) was Boc-protected before the peptide coupling step or after. Compound 20a was synthesized according to a reported procedure [15] and the carboxylic acid was selectively reduced with BH3∙SMe2 in dry THF to give the alcohol 21a in 95% yield. A Parikh-Doering oxidation [16] generated the aldehyde 22a (95%) and the subsequent addition of the Grignard reagent pent-4-enylmagnesium bromide furnished the alcohol 23a in 65% yield. This compound was oxidized to the corresponding ketone 24a in 76% yield using Dess-Martin periodinane in DCM [17]. The ester functionality in 24a was hydrolyzed using aqueous LiOH in dioxane and the resulting carboxylic acid was coupled with compound 19 using HATU and DIPEA in DMF to afford compound 25a in 89% yield over two steps. Compound 25b was synthesized in the same manner as 25a using commercially available 20b instead of 20a, and amine 18a instead of 19 in the coupling reaction. For purification reasons, the amino group in compound 25b was protected using Boc2O and TEA in MeOH to give compound 26 in 69% yield. A ring-closing metathesis protocol was performed on compound 25a and 26 using Hoveyda-Grubbs 2nd generation catalyst in refluxing DCM to furnish the macrocyclic compounds 27a and 27b in 21% and 47% yield, respectively. The cyclization resulted in exclusive formation of the trans isomers, as confirmed by 1H-NMR analyses. Finally, deprotection of the Boc group was achieved with Et3SiH and TFA in DCM to give the two target compounds 28a and 28b in 91% yield in both cases. Targets 34 and 35 (see Table 1) were synthesized according to the same synthetic route as 28a, using amine 8 instead of 19 and the methyl ester 24b instead of 24a for compound 35. Target 36 (see Table 1) was synthesized in the same manner as 28b using amine 18b instead of 18a.
When using the route described in Scheme 3, the ring-closing metathesis step was unsuccessful when trying to synthesize target 33 (see Table 1). The reason for this is at this stage unknown. Therefore, a different synthetic route was outlined for 33, as shown in Scheme 4. The methyl ester functionality in compound 24b was hydrolyzed using LiOH (aq.) in dioxane/H2O to furnish the corresponding carboxylic acid, which was subsequently used in a peptide coupling step with the amine (S)-2-(allyloxy)-1-((S)-2,2-dimethyl-1,3-dioxolan-4-yl)ethanamine [9] using HATU and DIPEA in DMF to give compound 29 in 57% yield. The acetal was hydrolyzed with 80% AcOH to generate the diol 30 in 95% yield. The ring-closing metathesis protocol, using Hoveyda-Grubbs 2nd generation catalyst in refluxing DCM, was now successful and rendered the macrocycle 31 in 88% yield. The primary hydroxyl group in 31 was tosylated by the use of TEA, Bu2SnO, and p-TsCl in DCM and DMF, which furnished compound 32 (57%). Finally, the tosylated compound was reacted with the amine 2-(5-(tert-butyl)oxazol-2-yl)propan-2-amine (see supp. info.) and DIPEA in refluxing EtOH to give the target compound 33 in 5% yield.
Structure Activity Relationships
All target compounds were screened against BACE-1, both in enzyme- and cell-based assays, and the IC50 values are shown in Table 1, along with Caco-2 papp values for selected compounds. The macrocycles were all 15-membered, except for compound 36 which was 16-membered.
Replacement of the terminal P2 amide functionality in the lead compound 1a [9] with a keto group resulted in target product 34 which showed retained potencies against BACE-1 although without displaying improved permeability. It has earlier been found that BACE-1 inhibitors with a sulfonamide substituted isophthalamide in the P2 position are substrates for P-gp efflux [18], which could explain the low permeability of compound 34. Replacement of the P2 sulfonamide functionality with a less polar methyl group furnished compound 35, resulting in a significantly improved (papp = 13 x 10-6 cm/s) permeability. However, with the P2 sulfonamide replaced with a methyl group the BACE-1 activity was substantially reduced, but this compound exhibited comparable BACE-1 activities as the amide analogue 1b [9] (Table 1) published earlier (a BACE-1 IC50 value of 1100 vs. 500 nM and a cell-based IC50 value of 1400 vs. 220 nM, respectively), but with significantly better permeability (a Caco-2 value of 13 x 10-6 cm/s, as mentioned above, vs. < 1 x 10-6 cm/s, respectively). In-house studies have shown that the introduction of a oxazole group (see supplementary material) in the P2´ position can give rise to highly active compounds and with improved permeability. Introduction of this oxazole group provided compound 33 which exhibited additional improvement in permeability compared with inhibitor 35 whilst retaining enzyme- and cell-based activity compared with compound 35. The X-ray structure of 33 (PDB id: 4GMI) in complex with BACE-1 shows that the gem-methyl groups shielding the secondary nitrogen atom are well accommodated for and, as such, are not exerting any steric impediment on compound binding (Fig. 2). Moreover, the P2’ tert-butyl substituted oxazole has a good fit to the S2’ subsite, including a hydrogen bond to Thr72 of the flap.
Generally, we have noted that replacing the macrocyclic ether oxygen with a carbon in these inhibitors often results in increased potency [19]. However, the carbon analogue to compound 34, inhibitor 28a, showed similar potent activities (a BACE-1 IC50 value of 8.9 vs. 12 nM and a cell-based IC50 value of 3.0 vs. 4.6 nM, respectively). As expected, with a P2 sulfonamide moiety, compound 28a also displayed a similar low Caco-2 permeability value (< 1 x 10-6 cm/s). As foreseen, the Boc-protected compound 27b was inactive. (It is well established that hydroxyethylamine-based BACE-1 inhibitors bind to the catalytic aspartates Asp32 and Asp228 via the protonated amino and hydroxy groups [9, 20, 21]). Compound 28b, incorporating a toluene core in the P2 position, displayed an improved potency compared to its analogue 35 (IC50 280 vs. 1100 nM, respectively) and equal permeability (papp = 13 x 10-6 cm/s). The 16-membered macrocycle 36 showed similar activity and permeability as its corresponding 15-membered macrocycle 28b. Attempts to synthesize the corresponding 14-membered macrocycle were unsuccessful, likely due to a high ring strain of the target product.
CONCLUSION
A novel series of macrocyclic BACE-1 inhibitors incorporating a P2/P3 keto functionality was investigated, with some inhibitors showing highly promising low nanomolar activities against the BACE-1 enzyme and in the cell-based assay. Moreover, several inhibitors exhibited improved cell-based permeability compared to inhibitors previously synthesized, but, not unexpectedly, these target compounds displayed decreased potency due to replacement of the sulfonamide functionality in the P2 position with a methyl group, where the sulfonamide moiety has been shown to be important for the activity in this type of inhibitor compounds. In summary, improved permeability could be achieved compared to earlier published inhibitors from our group whilst retaining the overall BACE-1 inhibitory activities.
Further work on this series is highly warranted exploring sulfonamide replacements in the P2-position, e.g. by use of small lipophilic groups and by introducing potency-enhancing lipophilic substituents protruding into the S3 sub pocket.
EXPERIMENTAL SECTION
General
NMR-spectra were recorded on a Varian 300 MHz instrument using CDCl3, CD3OD or DMSO-d6 as solvents. Thin layer chromatography (TLC) was carried out on Merck precoated 60 F254 plates using UV-light and charring with ethanol/sulfuric acid/p-anisaldehyde/acetic acid 90:3:2:1, or a solution of 0.5% ninhydrin in ethanol, for visualization. Flash column chromatography was performed using silica gel 60 (0.040-0.063 mm, Merck). Organic phases were dried over anhydrous magnesium sulfate. Optical rotations were measured using a Perkin-Elmer 141 polarimeter at 22 °C. Gradient LC-MS was performed on a Gilson system (column: Phenomenex C18, 100 x 21 mm, 5 µm for preparative runs and xBridgeTM C18, 50 x 4.6 mm, 2.5 ìm for analytical runs; pump: Gilson gradient pump 322; UV/VIS-detector: Gilson 152; MS detector: Thermo Finnigan Surveyor MSQ; Gilson Fraction Collector FC204) using acetonitrile with 0.05% formic acid and deionized water with 0.05% formic acid as mobile phases. High resolution mass spectra (HRMS) were recorded on a Waters Synapt HDMS instrument equipped with an electrospray interface.
BACE-1 Enzyme Assay
The BACE-1 assay was performed as previously described [22].
BACE-1 Cell Assay
The cell-based assay was performed as previously described [23].
Caco-2 Assay
The permeability coefficients (papp) were performed as previously described [24].
LC-MS Purity Measurements
Chromatography system A. Column: Phenomenex C18, 50 x 4.6 mm, 3 µm; pump: Gilson gradient pump 322; UV/VIS-detector: Gilson 152; MS detector: Thermo Finnigan Surveyor MSQ; Software: Gilson Unipoint 4.0 and Xcalibur 1.3. Mobile phase A: 10 mM NH4OAc in water; mobile phase B: 10 mM NH4OAc in 90% acetonitrile; gradient: 20-100% B over 6 min at 1 mL/min followed by 100% of B for 4 min at 1 mL/min. Peaks were detected at 254 nm.
Chromatography system B. Column: XBridge C8, 50 x 4.6 mm, 2.5 ìm; pump: Gilson gradient pump 322; UV/VIS-detector: Gilson 152; MS detector: Thermo Finnigan Surveyor MSQ; Software: Gilson Unipoint 4.0 and Xcalibur 1.3. Mobile phase A: 10 mM NH4OAc in water; mobile phase B: 10 mM NH4OAc in 90% acetonitrile; gradient: 40-100% B over 7 min at 1 mL/min followed by 100% of B for 3 min at 1 mL/min. Peaks were detected at 254 nm.
SYNTHETIC PROCEDURES
(2S,3S)-4-(Allyloxy)-3-azidobutane-1,2-diol (3). Compound 2 (1.82 g, 8.0 mmol) was dissolved in AcOH/H2O (40:15 mL) and the solution was stirred for 3 hours at 60 °C. The mixture was concentrated, co-evaporated with toluene and purified using flash column chromatography (toluene/EtOAc 1:1) to give compound 3 (1.17 g, 78%) as a colorless oil. 1H-NMR (CDCl3, 300 MHz): δ 2.73 (bs, 2H), 3.58–3.66 (m, 2H), 3.67-3.77 (m, 4H), 4.03 (d, J = 5.7 Hz, 2H), 5.19–5.32 (m, 2H), 5.82–5.95 (m, 1H); 13C-NMR (CDCl3, 75.5 MHz): δ 62.1, 63.6, 70.0, 71.9, 72.6, 117.9, 134.0. MS-analysis resulted in decomposition. No mass peak could be detected.
(2S,3S)-4-(Allyloxy)-3-azido-2-hydroxybutyl 4-methyl benzenesulfonate (4). TEA (830 ìL, 6.0 mmol), Bu2SnO (124 mg, 0.5 mmol) and p-TsCl (1.13 g, 5.9 mmol) were added to a solution of compound 3 (930 mg, 4.9 mmol) in dry DCM (30 mL). The mixture was stirred for 3.5 hours at room temperature. The solution was thereafter diluted with DCM, washed two times with brine and the organic phase was dried, filtered and concentrated. Purification using flash column chromatography (toluene/EtOAc 19:1) gave compound 4 (1.25 g, 74%) as a colorless oil. 1H-NMR (CDCl3, 300 MHz): δ 2.45 (s, 3H), 2.87 (bs, 1H), 3.55-3.61 (m, 1H), 3.66 (dd, J = 6.0, 10.2 Hz, 1H), 3.75 (dd, J = 3.6, 10.2 Hz, 1H), 3.83-3.90 (m, 1H), 4.01 (dt, J = 1.5, 5.7 Hz, 2H), 4.10 (dd, J = 5.7, 10.5 Hz, 1H), 4.20 (dd, J = 3.3, 10.5 Hz, 1H), 5.18-5.31 (m, 2H), 5.80-5.93 (m, 1H), 7.36 (d, J = 8.1 Hz, 2H), 7.81 (d, J = 8.1 Hz, 2H); 13C-NMR (CDCl3, 75.5 MHz): δ 21.7, 61.4, 69.5, 69.6, 71.1, 72.5, 117.7, 128.1, 130.1, 132.5, 134.0, 145.4 MS-analysis resulted in decomposition. No mass peak could be detected.
(S)-2-((S)-2-(Allyloxy)-1-azidoethyl)oxirane (5). Compound 4 (1.25 g, 3.7 mmol) was dissolved in MeOH (28 mL). K2CO3 (830 mg, 6.0 mmol) was added at 0 °C and the solution was left to reach room temperature and stirred for 3 hours. H2O and DCM were added and the organic phase was dried, filtered and concentrated. The crude material was purified using flash column chromatography (toluene/EtOAc 19:1) and compound 5 (521 mg, 84%) was collected as a colorless oil. 1H-NMR (CDCl3, 300 MHz): δ 2.75–2.77 (m, 1H), 2.79-2.83 (m, 1H), 3.06–3.10 (m, 1H), 3.46–3.51 (m, 1H), 3.58 (dd, J = 6.3, 9.9 Hz, 1H), 3.66 (dd, J = 3.9, 9.9 Hz, 1H), 4.03 (dt, J = 1.5, 5.7 Hz, 2H), 5.18-5.21 (m, 1H), 5.24-5.31 (m, 1H), 5.83–5.96 (m, 1H); 13C-NMR (CDCl3, 75.5 MHz): δ 45.2, 50.8, 61.8, 70.0, 72.5, 117.6, 134.1 MS (2M+Na)+ calcd: 361.2; found: 360.7.
(2R,3S)-4-(Allyloxy)-3-azido-1-((3-isopropylbenzyl) amino)butan-2-ol (6). Compound 5 (385 mg, 2.3 mmol) and 3-isopropylbenzylamine (510 mg, 3.4 mmol) were dissolved in MeOH (2.5 mL). TEA (640 mL, 4.6 mmol) was added and the solution was stirred for 44 hours at 30 °C. The mixture was concentrated and purified using flash column chromatography (toluene/EtOAc 3:1 + 1% TEA) to give compound 6 (405 mg, 56%) as a slightly yellow oil. 1H-NMR (CDCl3, 300 MHz): δ 1.25 (d, J = 6.9 Hz, 6H), 2.70 (dd, J = 8.1, 12.3 Hz, 1H), 2.83-2.94 (m, 2H), 3.52-3.57 (m, 1H), 3.58-3.64 (m, 1H), 3.65-3.71 (m, 1H), 3.74 (dd, J = 3.6, 9.9 Hz, 1H), 3.77 (d, J = 3.6 Hz, 2H), 4.02 (dt, J = 1.5, 5.7 Hz, 2H), 5.17–5.32 (m, 2H), 5.83–5.96 (m, 1H), 7.09–7.18 (m, 3H), 7.22-7.28 (m, 1H); 13C-NMR (CDCl3, 75.5 MHz): δ 24.0, 34.1, 50.9, 53.8, 63.9, 68.9, 70.1, 72.3, 117.3, 125.3, 125.5, 126.2, 128.5, 134.2, 139.7, 149.2 MS (M+H)+ calcd: 319.2; found: 319.6.
tert-Butyl ((2R,3S)-4-(allyloxy)-3-azido-2-hydroxybutyl) (3-isopropyl-benzyl)carbamate (7). Compound 6 (228 mg, 0.7 mmol) was dissolved in MeOH (7 mL). TEA (200 ìL, 1.4 mmol) and Boc2O (229 mg, 1.0 mmol) were added and the mixture was stirred for 1 h. The solution was concentrated and purified using flash column chromatography (toluene/EtOAc 19:1), which gave compound 7 (243 mg, 81%) as a colorless oil. 1H-NMR (CDCl3, 300 MHz): δ 1.25 (d, J = 6.9 Hz, 6H), 1.47 (s, 9H), 2.85-2.94 (m, 1H), 3.36 (dd, J = 2.4, 14.7 Hz, 1H), 3.45 (td, J = 3.3, 7.2 Hz, 1H), 3.63 (dd, J = 7.2, 10.2 Hz, 1H), 3.72 (bs, 1H), 3.79 (bs, 1H), 4.02 (dt, J = 1.5, 5.4 Hz, 2H), 4.45 (bs, 2H), 4.63 (bs, 1H), 5.17-5.22 (m, 1H), 5.25-5.32 (m, 1H), 5.83-5.96 (m, 1H), 7.09 (s, 1H), 7.13-7.18 (m, 2H), 7.23-7.24 (m, 1H); 13C-NMR (CDCl3, 75.5 MHz): δ 24.0, 28.4, 34.1, 51.1, 52.8, 63.4, 70.1, 71.6, 72.3, 79.1, 117.3, 124.9, 125.3, 126.2, 129.0, 134.3, 137.7, 149.4, 153.1 MS (M+H)+ calcd: 419.3; found: 419.5.
tert-Butyl ((2R,3S)-4-(allyloxy)-3-amino-2-hydroxybutyl) (3-isopropyl-benzyl)carbamate (8). Compound 7 (114 mg, 0.3 mmol) was dissolved in MeOH (3 mL). Aliquat 336 (1 drop), TEA (190 ìL, 1.3 mmol) and 1,3-propanedithiol (135 ìL, 1.3 mmol) were added to the solution and the mixture was stirred for 6 days at room temperature. The reaction mixture was filtered, concentrated and purified using flash column chromatography (MeOH/EtOAc 1:9 + 1% TEA) to give compound 8 (82 mg, 76%) as a colorless oil. 1H-NMR (CDCl3, 300 MHz): δ 1.24 (d, J = 6.9 Hz, 6H), 1.46 (s, 9H), 2.84-2.93 (m, 3H), 2.96–3.02 (m, 1H), 3.35-3.39 (m, 1H), 3.44-3.49 (m, 2H), 3.55–3.57 (m, 1H), 3.75-3.85 (m, 1H), 3.96-3.98 (m, 2H), 4.50 (bs, 2H), 5.14–5.17 (m, 1H), 5.21-5.28 (m, 1H), 5.81–5.94 (m, 1H), 7.02–7.18 (m, 3H), 7.21-7.27 (m, 1H); 13C-NMR (CDCl3, 75.5 MHz): δ 24.1, 28.5, 34.2, 50.4, 52.4, 53.8, 71.8, 72.3, 73.6, 80.7, 117.3, 124.9, 125.4, 128.6, 134.6, 138.2, 149.3, 153.2 MS (M+H)+ calcd: 393.3; found: 393.6.
(S)-1-((R)-2,2-Dimethyl-1,3-dioxolan-4-yl)hex-5-en-1-ol (9a). Magnesium turnings (1.71 g, 70.3 mmol), a crystal of iodine and dry THF (30 mL) were mixed in a two-necked round-bottom flask equipped with a cooler. The mixture was heated gently and 5-bromopent-1-ene (8.3 mL, 70.2 mmol) was added slowly to maintain a steady reflux. After the addition was complete, 30 mL of dry THF was added, and the solution was refluxed for 1 h. The resulting Grignard mixture was thereafter cooled to -30 °C and isopropylidene-protected D-glyceraldehyde (6.03 g, 46.4 mmol) dissolved in dry THF (50 mL) was added dropwise over a period of 30 min. The resulting solution was allowed to obtain room temperature overnight, after which Et2O and saturated NH4Cl were added. The water phase was extracted with Et2O twice and the combined organic phases were dried, filtered and evaporated. The crude material was purified using flash column chromatography (heptane/EtOAc 10:1) to yield 9a (6.31 g, 68%) as a colorless oil. (S,R): 1H-NMR (CDCl3, 300 MHz): δ 1.31-1.38 (m, 1H), 1.36 (s, 3H), 1.39-1.54 (m, 2H), 1.42 (s, 3H), 1.57-1.68 (m, 1H), 2.04-2.13 (m, 2H), 2.15 (d, J = 3.5 Hz, 1H), 3.71-3.80 (m, 1H), 3.86-4.05 (m, 3H), 4.93-5.06 (m, 2H), 5.73-5.88 (m, 1H); 13C-NMR (CDCl3, 75.5 MHz): δ 25.1, 25.4, 26.6, 32.2, 33.7, 64.7, 70.7, 78.8, 109.0, 114.9, 138.5.
1-((R)-2,2-Dimethyl-1,3-dioxolan-4-yl)hept-6-en-1-ol (9b). Compound 9b (9.62 g, total yield 87%, (S)/(R) 75:25) was synthesized from isopropylidene-protected D-glyceraldehyde in the same manner as 9a. The two diastereomers were separated and collected as yellow oils. (S,R): 1H-NMR (CDCl3, 300 MHz): δ 1.31-1.64 (m, overlapped, 6H), 1.37 (s, overlapped, 3H), 1.43 (s, overlapped, 3H), 1.93 (d, J = 3.0 Hz, 1H), 2.07 (app. q, J = 6.6 Hz, 2H), 3.76-3.81 (m, 1H), 3.88-3.97 (m, 2H), 3.99-4.06 (m, 1H), 4.93-5.04 (m, 2H), 5.74-5.87 (m, 1H); 13C-NMR (CDCl3, 75.5 MHz): δ 25.4, 25.5, 26.6, 29.0, 32.6, 33.8, 64.6, 70.8, 78.8, 109.1, 114.6, 138.9 MS (M+H)+ calcd: 214.2; found: 213.9 (R,R): 1H-NMR (CDCl3, 300 MHz): δ 1.29-1.63 (m, overlapped, 6H), 1.37 (s, overlapped, 3H), 1.44 (s, overlapped, 3H), 2.04-2.10 (m, 2H), 2.12 (d, J = 5.1 Hz, 1H), 3.45-3.53 (m, 1H), 3.69-3.77 (m, 1H), 3.95-4.04 (m, 2H), 4.93-5.03 (m, 2H), 5.74-5.88 (m, 1H); 13C-NMR (CDCl3, 75.5 MHz): δ 25.2, 25.5, 26.8, 29.0, 33.7, 33.8, 66.3, 72.4, 79.3, 109.5, 114.6, 138.9 MS (M+H)+ calcd: 214.2; found: 213.8.
(R)-1-((R)-2,2-Dimethyl-1,3-dioxolan-4-yl)hex-5-en-1-yl 4-nitrobenzoate (10a). To a solution of 9a (1.63 g, 8.15 mmol) in dry THF (50 mL), p-nitrobenzoic acid (2.23 g, 13.3 mmol) and Ph3P (3.42 g, 13.0 mmol) were added. DIAD (2.57 mL, 13.1 mmol) was added dropwise over a period of 30 min and the resulting reaction mixture was stirred overnight at room temperature. After evaporation of the solvent, the crude remainder was purified by flash column chromatography (toluene → toluene/EtOAc 18:1) to furnish compound 10a (2.60 g, 91%) as a slightly yellow oil. 1H-NMR (CDCl3, 300 MHz): δ 1.35 (s, 3H), 1.42-1.56 (m, 2H), 1.45 (s, 3H), 1.66-1.85 (m, 2H), 2.03-2.19 (m, 2H), 3.79 (dd, J = 6.0, 8.6 Hz, 1H), 4.03-4.12 (m, 1H), 4.30 (dd, J = 6.0, 11.9 Hz, 1H), 4.92-5.06 (m, 2H), 5.25 (dt, J = 4.9, 9.9 Hz, 1H), 5.69-5.85 (m, 1H), 8.23 (d, J = 9.2 Hz, 2H), 8.29 (d, J = 9.2 Hz, 2H); 13C-NMR (CDCl3, 75.5 MHz): δ 24.6, 25.3, 26.4, 30.2, 33.4, 65.8, 75.3, 76.6, 109.8, 115.2, 123.6, 130.9, 135.7, 138.0, 150.7, 164.5 MS (M+H)+ calcd: 350.2; found: 350.5.
(R)-1-((R)-2,2-Dimethyl-1,3-dioxolan-4-yl)hex-5-en-1-ol (11a). Compound 10a (2.55 g, 7.31 mmol) was dissolved in MeOH (40 mL) and NaOMe in MeOH (0.5 M, 15 mL, 7.5 mmol) was added dropwise during 20 min. The solution was stirred at room temperature for 1 h and 45 min, after which it was neutralized with HOAc and evaporated. DCM and saturated NaHCO3 were added and the water phase was extracted with DCM once. The organic phases were dried, filtered and evaporated and the crude material was purified using flash column chromatography (heptane/EtOAc 9:1) to yield 11a (1.00 g, 68%) as a colorless oil. 1H-NMR (CDCl3, 300 MHz): δ 1.32-1.56 (m, 3H), 1.36 (s, 3H), 1.43 (s, 3H), 1.57-1.72 (m, 1H), 2.01-2.14 (m, 2H), 2.42 (d, J = 4.7 Hz, 1H), 3.42-3.54 (m, 1H), 3.66-3.76 (m, 1H), 3.93-4.04 (m, 2H), 4.92-5.06 (m, 2H), 5.73-5.87 (m, 1H); 13C-NMR (CDCl3, 75.5 MHz): δ 24.8, 25.4, 26.7, 33.0, 33.6, 66.2, 72.1, 79.3, 109.4, 114.8, 138.5
(S)-4-((S)-1-Azidohex-5-en-1-yl)-2,2-dimethyl-1,3-dioxolane (12a). To a solution of 11a (993 mg, 4.97 mmol) in dry THF (30 mL), Ph3P (2.06 g, 7.85 mmol) was added and the temperature was lowered to -5 °C. DIAD (1.60 mL, 8.13 mmol) and DPPA (2.17 g, 7.89 mmol) were added dropwise during 20 min and 5 min, respectively, and the stirred solution was allowed to reach room temperature overnight. After evaporation of the solvent the crude mixture was purified using flash column chromatography (2.5% EtOAc in heptane) to give 12a (1.10 g, 98%) as a slightly yellow oil. 1H-NMR (CDCl3, 300 MHz): δ 1.28-1.46 (m, 1H), 1.35 (s, 3H), 1.45 (s, 3H), 1.47-1.70 (m, 3H), 2.04-2.14 (m, 2H), 3.44-3.53 (m, 1H), 3.83-3.90 (m, 1H), 3.98-4.10 (m, 2H), 4.95-5.08 (m, 2H), 5.72-5.86 (m, 1H); 13C-NMR (CDCl3, 75.5 MHz): δ 25.3, 25.5, 26.3, 30.4, 33.4, 63.7, 66.0, 78.0, 109.7, 115.2, 138.1 MS (M+H)+ calcd: 226.2; found: 226.3.
(S)-4-((S)-1-Azidohept-6-en-1-yl)-2,2-dimethyl-1,3-dioxolane (12b). Compound 12b (357 mg, 88%) was synthesized from (R,R)-9b in the same manner as 12a and was collected as a colorless oil. 1H-NMR (CDCl3, 300 MHz): δ 1.34-1.47 (m, overlapped, 6H), 1.36 (s, overlapped, 3H), 1.46 (s, overlapped, 3H), 1.53-1.58 (m, 2H), 3.45-3.49 (m, 1H), 3.84-3.91 (m, 1H), 3.99-4.09 (m, 2H), 4.93-5.05 (m, 2H), 5.73-5.87 (m, 1H); 13C-NMR (CDCl3, 75.5 MHz): δ 25.4, 25.8, 26.4, 28.7, 31.0, 33.6, 64.0, 66.1, 78.0, 109.8, 114.8, 138.6 MS (M+H)+ calcd: 240.2; found: 239.9.
(2S,3S)-3-Azidooct-7-ene-1,2-diol (13a). Compound 12a (1.07 g, 4.76 mmol) was dissolved in a mixture of HOAc (20 mL) and H2O (10 mL). The solution was stirred at 60 °C for 3 h after which the solvents were evaporated. The crude material was purified using flash column chromatography (toluene/EtOAc 2:1) to yield 13a (650 mg, 74%) as a colorless oil. 1H-NMR (CDCl3, 300 MHz): δ 1.40-1.72 (m, 4H), 2.02-2.18 (m, 2H), 3.36-3.46 (m, 1H), 3.57-3.75 (m, 3H), 3.85 (br s, 1H), 4.10 (br s, 1H), 4.95-5.08 (m, 2H), 5.72-5.87 (m, 1H); 13C-NMR (CDCl3, 75.5 MHz): δ 25.5, 29.8, 33.3, 63.2, 64.4, 73.9, 115.1, 138.0. MS-analysis resulted in decomposition. No mass peak could be detected.
(2S,3S)-3-Azidonon-8-ene-1,2-diol (13b). Compound 13b (290 mg, quant.) was synthesized from 12b in the same manner as 13a and was collected as a colorless oil. 1H-NMR (CDCl3, 300 MHz): δ 1.41-1.49 (m, 2H), 1.50-1.57 (m, 2H), 1.58-1.68 (m, 2H), 2.04-2.12 (m, 2H), 3.42-3.48 (m, 1H), 3.67-3.69 (m, 1H), 3.71-3.75 (m, 2H), 4.94-5.05 (m, 2H), 5.74-5.87 (m, 1H); 13C-NMR (CDCl3, 75.5 MHz): δ 25.8, 28.8, 30.5, 33.6, 63.3, 64.8, 73.6, 114.8, 138.6 MS-analysis resulted in decomposition. No mass peak could be detected.
(2S,3S)-3-Azido-2-hydroxyoct-7-en-1-yl 4-methyl benzenesulfonate (14a). To a solution of 13a (588 mg, 3.18 mmol) in a mixture of DCM (50 mL) and DMF (10 mL), TEA (885 µL, 6.35 mmol), Bu2SnO (81 mg, 0.33 mmol) and TsCl (750 mg, 3.93 mmol) were added. The reaction mixture was stirred at room temperature overnight. DCM and brine were added, and the organic phase was dried, filtered and evaporated. Flash column chromatography (toluene/EtOAc 9:1) afforded compound 14a (900 mg, 84%) as a colorless oil. 1H-NMR (CDCl3, 300 MHz): δ 1.38-1.70 (m, 4H), 2.01-2.14 (m, 2H), 2.45 (s, 3H), 2.64 (d, J = 5.0 Hz, 1H), 3.35-3.46 (m, 1H), 3.75-3.85 (m, 1H), 4.07 (dd, J = 6.6, 10.4 Hz, 1H), 4.17 (dd, J = 3.1, 10.4 Hz, 1H), 4.94-5-06 (m, 2H), 5.70-5.85 (m, 1H), 7.37 (d, J = 8.2 Hz, 2H), 2.83 (d, J = 8.2 Hz, 2H); 13C-NMR (CDCl3, 75.5 MHz): δ 21.8, 25.3, 29.8, 33.4, 63.6, 71.1, 71.6, 115.3, 128.1, 130.1, 132.5, 138.0, 145.5 MS-analysis resulted in decomposition. No mass peak could be detected.
(2S,3S)-3-Azido-2-hydroxynon-8-en-1-yl 4-methyl benzenesulfonate (14b). Compound 14b (395 mg, 88%) was synthesized from 13b in the same manner as 14a and was collected as a pale yellow oil. 1H-NMR (CDCl3, 300 MHz): δ 1.36-1.46 (m, 3H), 1.49-1.57 (m, 2H), 1.60-1.68 (m, 1H), 2.03-2.10 (m, 2H), 2.36-2.38 (m, 1H), 2.46 (s, 3H), 3.38-3.44 (m, 1H), 3.76-3.84 (m, 1H), 4.08 (dd, J = 6.6, 10.5 Hz, 1H), 4.19 (dd, J = 3.0, 10.5 Hz, 1H), 4.94-5.05 (m, 2H), 5.72-5.86 (m, 1H), 7.37 (d, J = 8.4 Hz, 2H), 7.81 (d, J = 8.4 Hz, 2H); 13C-NMR (CDCl3, 75.5 MHz): δ 21.8, 25.5, 28.7, 33.6, 63.7, 71.1, 71.7, 114.9, 128.2, 130.2, 132.6, 138.5, 145.5 MS-analysis resulted in decomposition. No mass peak could be detected.
(S)-2-((S)-1-Azidohex-5-en-1-yl)oxirane (15a). Compound 14a (320 mg, 0.94 mmol) was dissolved in MeOH (10 mL) and the temperature of the solution was lowered to 0 °C. Na2CO3 (175 mg, 1.65 mmol) was added and the stirred mixture was allowed to obtain room temperature overnight. Saturated NH4Cl was added and the resulting mixture was extracted with DCM. The organic phase was dried, filtered and evaporated and the crude remainder was purified using flash column chromatography (toluene) to provide the somewhat volatile 15a (157 mg, 100%) as a colorless oil. 1H-NMR (CDCl3, 300 MHz): δ 1.43-1.72 (m, 4H), 2.08 (q, J = 6.6 Hz, 2H), 2.72-2.79 (m, 2H), 2.94-3.00 (m, 1H), 3.26-3.34 (m, 1H), 4.94-5.08 (m, 2H), 5.71-5.86 (m, 1H); 13C-NMR (CDCl3, 75.5 MHz): δ 24.9, 31.3, 33.4, 44.8, 53.4, 62.5, 115.1, 138.0. MS (2M+H)+ calcd: 335.2; found: 335.6.
(S)-2-((S)-1-Azidohept-6-en-1-yl)oxirane (15b). Compound 15b (192 mg, 97%) was synthesized from 14b in the same manner as 15a and was collected as a pale yellow oil. 1H-NMR (CDCl3, 300 MHz): δ 1.40-1.48 (m, 2H), 1.50-1.60 (m, 2H), 1.62-1.72 (m, 2H), 2.04-2.12 (m, 2H), 2.76-2.79 (m, 1H), 2.80-2.83 (m, 1H), 2.99-3.03 (m, 1H), 3.31 (dt, J = 5.1, 8.1 Hz, 1H), 4.94-5.05 (m, 2H), 5.74-5.87 (m, 1H); 13C-NMR (CDCl3, 75.5 MHz): δ 25.3, 28.7, 31.9, 33.6, 45.0, 53.6, 62.8, 114.8, 138.6 MS (2M+H)+ calcd: 363.2; found: 362.9.
(2R,3S)-3-Azido-1-((3-isopropylbenzyl)amino)oct-7-en-2-ol (16a). Compound 15a (112 mg, 0.67 mmol) was dissolved in MeOH (1 mL) and 3-isopropylbenzylamine (145 mg, 0.97 mmol) and TEA (185 µL, 1.33 mmol) were added. The solution was stirred overnight at 30 °C and was then evaporated. The crude material was purified using flash column chromatography (toluene/EtOAc 3:1 → toluene/EtOAc 1:1 + 1% TEA), yielding compound 16a (130 mg, 61%) as a colorless oil. 1H-NMR (CDCl3, 300 MHz): δ 1.25 (d, J = 6.9 Hz, 6H), 1.34-1.80 (m, 4H), 1.98-2.14 (m, 2H), 2.68 (dd, J = 8.5, 12.2 Hz, 1H), 2.81 (dd, J = 3.6, 12.2 Hz, 1H), 2.84-2.96 (m, 2H), 3.02 (bs, 2H), 3.32-3.42 (m, 1H), 3.58-3.67 (m, 1H), 3.72 (d, J = 13.0 Hz, 1H), 3.80 (d, J = 13.0 Hz, 1H), 4.92-5.07 (m, 2H), 5.70-5.86 (m, 1H), 7.07-7.17 (m, 2H), 7.21-7.29 (m, 1H); 13C-NMR (CDCl3, 75.5 MHz): δ 24.1, 25.7, 29.9, 33.5, 34.1, 50.3, 53.9, 65.7, 71.4, 115.1, 125.4, 125.6, 126.4, 128.6, 138.2, 139.6, 149.3 MS (M+H)+ calcd: 317.2; found: 317.6.
(2R,3S)-3-Azido-1-((3-isopropylbenzyl)amino)non-8-en-2-ol (16b). Compound 16b (177 mg, 53%) was synthesized from 15b in the same manner as 16a and was collected as a colorless oil. 1H-NMR (CDCl3, 300 MHz): δ 1.26 (d, J = 6.9 Hz, 6H), 1.37-1.50 (m, 4H), 1.52-1.66 (m, 2H), 2.04-2.11 (m, 2H), 2.71 (dd, J = 8.7, 12.3 Hz, 1H), 2.85 (dd, J = 3.6, 12.3 Hz, 1H), 2.87-2.96 (m, 1H), 3.36-3.42 (m, 1H), 3.60-3.66 (m, 1H), 3.79 (d, J = 4.2 Hz, 2H), 4.94-5.05 (m, 2H), 5.74-5.88 (m, 1H), 7.11-7.16 (m, 3H), 7.25-7.30 (m, 1H); 13C-NMR (CDCl3, 75.5 MHz): δ 24.1, 26.0, 28.8, 30.5, 33.7, 34.2, 50.2, 54.0, 65.8, 71.4, 114.7, 125.5, 125.6, 126.4, 128.7, 138.7, 139.8, 149.4 MS (M+H)+ calcd: 331.2; found: 330.9.
tert-Butyl ((2R,3S)-3-azido-2-hydroxyoct-7-en-1-yl)(3-isopropylbenzyl)carbamate (17). Compound 17 (193 mg, 97%) was synthesized from 16a according to the synthesis of compound 7 and was collected as a colorless oil. 1H-NMR (CDCl3, 300 MHz): δ 1.25 (d, J = 6.9 Hz, 6H), 1.49 (s, 9H), 1.53-1.63 (m, 4H), 2.05-2.10 (m, 2H), 2.85-2.94 (m, 1H), 3.19-3.25 (m, 2H), 3.50-3.60 (m, 1H), 3.65-3.75 (m, 1H), 4.45 (bs, 2H), 4.94-5.05 (m, 2H), 5.71-5.85 (m, 1H), 7.04 (d, J = 7.5 Hz, 1H), 7.09 (s, 1H), 7.14-7.19 (m, 1H), 7.24-7.29 (m, 1H); 13C-NMR (CDCl3, 75.5 MHz): δ 24.1, 25.6, 28.5, 29.7, 33.6, 34.2, 52.9, 54.9, 65.4, 74.2, 81.4, 115.2, 125.1, 125.9, 128.8, 137.8, 138.2, 149.6, 153.3 MS (M+H)+ calcd: 417.2; found: 417.0.
(2R,3S)-3-Amino-1-((3-isopropylbenzyl)amino)oct-7-en-2-ol (18a). To a solution of 16a (115 mg, 0.36 mmol) in MeOH (1.5 mL), TEA (250 µL, 1.79 mmol), 1,3-propanedithiol (180 µL, 1.79 mmol) and Aliquat 336 (2 drops) were added. The solution was stirred at room temperature for 5 days, after which 1 M HCl, H2O and DCM were added. The aqueous phase was washed with DCM and was then made basic with 1 M NaOH. Subsequently, the water phase was extracted with EtOAc and the organic phase was dried, filtered and evaporated to yield 18a (88 mg, 83%) as a colorless oil. 1H-NMR (CD3OD, 300 MHz): δ 1.24 (d, J = 6.9 Hz, 6H), 1.27-1.60 (m, 4H), 2.01-2.12 (m, 2H), 2.59-2.71 (m, 2H), 2.72-2.83 (m, 1H), 2.83-2.95 (m, 2H), 3.59-3.68 (m, 1H), 3.73 (d, J = 13.0 Hz, 1H), 3.80 (d, J = 13.0 Hz, 1H), 4.88-5.06 (m, 2H), 5.72-5.90 (m, 1H), 7.06-7.28 (m, 3H); 13C-NMR (CD3OD, 75.5 MHz): δ 24.5, 26.6, 32.7, 34.8, 35.3, 51.3, 54.6, 56.0, 73.6, 115.2, 126.3, 127.0, 127.6, 129.5, 139.7, 140.5, 150.4 MS (M+H)+ calcd: 291.2; found: 291.6.
(2R,3S)-3-Amino-1-((3-isopropylbenzyl)amino)non-8-en-2-ol (18b). Compound 18b (128 mg, 81%) was synthesized in the same manner as 18a from 16b and was collected as a colorless oil. 1H-NMR (CDCl3, 300 MHz): δ 1.24 (d, J = 6.9 Hz, 6H), 1.27-1.45 (m, 6H), 2.01-2.08 (m, 2H), 2.43 (bs, 3H), 2.67 (dd, J = 8.7, 12.0 Hz, 1H), 2.79 (dd, J = 3.6, 12.0 Hz, 1H), 2.85-2.94 (m, 2H), 3.54-3.59 (m, 1H), 3.74 (d, J = 12.9 Hz, 1H), 3.82 (d, J = 12.9 Hz, 1H), 4.91-5.02 (m, 2H), 5.72-5.85 (m, 1H), 7.11-7.16 (m, 3H), 7.22-7.27 (m, 1H); 13C-NMR (CDCl3, 75.5 MHz): δ 24.1, 26.0, 29.0, 33.1, 33.7, 34.2, 50.0, 54.0, 54.5, 72.0, 114.6, 125.4, 125.7, 126.5, 128.6, 138.9, 139.7, 149.3 MS (M+H)+ calcd: 305.3; found: 305.4.
tert-Butyl ((2R,3S)-3-amino-2-hydroxyoct-7-en-1-yl) (3-isopropylbenzyl)carbamate (19). Compound 19 (151 mg, 88%) was synthesized in the same manner as 18a from 17 and was collected as a colorless oil. 1H-NMR (CD3OD, 300 MHz): δ 1.23 (d, J = 6.9 Hz, 6H), 1.27-1.60 (m, 13H), 2.03-2.10 (m, 2H), 2.67-2.72 (m, 1H), 2.82-2.92 (m, 1H), 3.00-3.10 (m, 1H), 3.35-3.42 (m, 1H), 3.65-3.80 (m, 1H), 4.35-4.50 (m, 1H), 4.55-4.65 (m, 1H), 4.89-5.02 (m, 2H), 5.74-5.87 (m, 1H), 7.03 (d, J = 7.5 Hz, 1H), 7.10.7.14 (m, 2H), 7.20.7.25 (m, 1H); 13C-NMR (CD3OD, 75.5 MHz): δ 24.5, 26.7, 28.8, 32.7, 34.9, 35.3, 50.0, 53.1, 55.4, 74.7, 81.3, 115.1, 125.8, 126.3, 129.5, 139.7, 150.3, 158.0. MS (M+H)+ calcd: 391.3; found: 391.0.
Methyl 3-(hydroxymethyl)-5-(N-methylmethylsulfo-namido)benzoate (21a). A solution of 20a (1.65 g, 5.75 mmol) in THF (30 ml, dry) was flushed with Ar at 0°C. After 10 minutes BH3SMe2 (930 ìL, 9.81 mmol) was added slowly and the mixture was left to reach room temperature and was stirred under Ar for 24 hours. The mixture was quenched with MeOH, concentrated, re-dissolved in EtOAc, washed with NaHCO3 (sat. aq.) and brine. The organic phase was dried, filtered and concentrated. Finally, purification using flash column chromatography (toluene/EtOAc 3:1) gave compound 21a (1.49 g, 95%) as white crystals. 1H-NMR (CD3OD, 300 MHz): δ 2.90 (s, 3H), 3.33 (s, 3H), 3.90 (s, 3H), 4.66 (s, 2H), 7.33 (s, 1H), 7.65 (s, 1H), 7.94 (s, 1H); 13C-NMR (CD3OD, 75.5 MHz): δ 35.6, 38.4, 53.8, 64.1, 126.9, 127.3, 130.1, 132.5, 143.6, 145.1, 167.7.
Methyl 3-(hydroxymethyl)-5-methylbenzoate (21b). Compound 21b (6.8 g, 69%) was synthesized from 20b in the same manner as 21a and was collected as a colorless oil. 1H-NMR (CDCI3, 400MHz) δ 1.80 (t, J = 5.9 Hz, 1H), 2.41 (s, 3H), 3.91 (s, 3H), 4.70 (d, J = 5.9 Hz, 2H), 7.40 (s, 1 H), 7.79 (s, 1 H), 7.83 (s, 1 H).
Methyl 3-formyl-5-(N-methylmethylsulfonamido) benzoate (22a). A mixture of SO3-pyridine complex (2.65 g, 16.6 mmol) in dry DMSO (8 mL) was added to a solution of 21a (1.42 g, 5.20 mmol) and TEA (8.7 mL, 62.4 mmol) in DMSO (17 mL) and the mixture was stirred for 90 minutes at room temperature. Water was added and the product was extracted into EtOAc three times. The combined organic extracts were dried, filtered and concentrated. Finally, purification using flash column chromatography (toluene/EtOAc 3:1) gave compound 22a (1.18 g, 84%) as a yellowish solid. 1H-NMR (CDCl3, 300 MHz): δ 2.91 (s, 3H), 3.41 (s, 3H), 3.98 (s, 3H), 8.09 (s, 1H), 8.28 (s, 1H), 8.43 (s, 1H), 10.06 (s, 1H); 13C-NMR (CDCl3, 75.5 MHz): δ 36.0, 37.9, 52.8, 129.4, 129.8, 131.8, 132.6, 137.6, 143.0, 165.2, 190.5.
Methyl 3-formyl-5-methylbenzoate (22b). Compound 22b (4.9 g, 73%) was synthesized from 21b in the same manner as 22a and was collected as a white solid. 1H-NMR (CDCl3, 300 MHz): δ 2.49 (s, 3H), 4.00 (s, 3H), 7.89 (s, 1H), 8.11 (s, 1H), 8.32 (s, 1H), 10.04 (s, 1H); 13C-NMR (CDCl3, 75.5 MHz): δ 21.2, 52.6, 129.0, 131.3, 133.5, 136.0, 136.8, 139.6, 163.5, 191.7.
Methyl 3-(1-hydroxyhex-5-en-1-yl)-5-(N-methyl methylsulfonamido) benzoate (23a). Freshly made pent-4-enylmagnesium bromide in THF (720 ìL, 0.65 mmol, 0.9 M) was added slowly to a solution of 22a (160 mg, 0.59 mmol) in dry Et2O (20 mL) and the mixture was stirred for 3 h at 0 °C. The reaction was quenched with NH4Cl (sat. aq.) and the product was extracted into EtOAc three times. The combined organic extracts were dried, filtered and concentrated. Finally, purification using flash column chromatography (toluene/EtOAc 3:1) gave compound 23a (130 mg, 65%) as a colorless oil. 1H-NMR (CDCl3, 300 MHz): δ 1.66–1.81 (m, 4H), 2.05-2.12 (m, 2H), 2.23 (bs, 1H), 2.85 (s, 3H), 3.35 (s, 3H), 3.93 (s, 3H), 4.75 (t, J = 6.4 Hz, 1H), 4.93–5.03 (m, 2H), 5.71–5.84 (m, 1H), 7.61 (s, 1H), 7.88 (s, 1H), 7.94 (s, 1H); 13C-NMR (CDCl3, 75.5 MHz): δ 25.0, 33.5, 35.7, 38.2, 38.7, 52.5, 73.6, 115.0, 125.1, 126.0, 129.1, 131.6, 138.4, 142.0, 147.1, 166.3 MS (M+Na)+ calcd: 364.1; found: 364.6.
Methyl 3-(1-hydroxyhex-5-en-1-yl)-5-methylbenzoate (23b). Compound 23b (580 mg, 79%) was synthesized from 22b in the same manner as 23a and was collected as a colorless oil. 1H-NMR (CDCl3, 300 MHz): δ 1.34-1.59 (m, 2H), 1.67-1.80 (m, 2H), 1.83-1.84 (m, 1H), 2.04-2.12 (m, 2H), 2.04 (s, 3H), 3.91 (s, 3H), 4.68-4.73 (m, 1H), 4.92-5.03 (m, 2H), 5.71-5.85 (m, 1H), 7.37 (s, 1H), 7.77 (s, 1H), 7.80 (s, 1H); 13C-NMR (CDCl3, 75.5 MHz): δ 21.4, 25.2, 33.7, 38.7, 52.2, 74.3, 114.9, 124.4, 129.5, 130.4, 131.3, 132.5, 138.6, 145.3, 164.4 MS (M+H)+ calcd: 249.1; found: 249.5.
Methyl 3-(hex-5-enoyl)-5-(N-methylmethylsulfonamido) benzoate (24a). Dess-Martin periodinane (248 mg, 0.59 mmol) was added to a solution of 23a (133 mg, 0.39 mmol) in DCM (3 mL) and the solution was stirred for 24 h at room temperature. The solution was diluted with Et2O and washed with Na2S2O3 and NaHCO3 (sat.). The organic phase was dried, filtered and concentrated. Purification using flash column chromatography (toluene/EtOAc 39:1) gave 24a (100 mg, 76%) as a colorless oil. 1H-NMR (CDCl3, 300 MHz): δ 1.81-1.91 (m, 2H), 2.13-2.20 (m, 2H), 2.87 (s, 3H), 3.01 (t, J = 7.5 Hz, 2H), 3.38 (s, 3H), 3.96 (s, 3H), 4.99-5.09 (m, 2H), 5.75-5.89 (m, 1H), 7.95 (s, 1H), 8.04 (s, 1H), 8.38 (s, 1H); 13C-NMR (CDCl3, 75.5 MHz): δ 21.6, 23.2, 33.2, 35.9, 38.0, 52.8, 115.7, 125.4, 127.8, 128.4, 129.2, 130.8, 138.0, 142.6, 165.7, 198.6 MS (M+H)+ calcd: 340.1; found: 340.6.
Methyl 3-(hex-5-enoyl)-5-methylbenzoate (24b). Compound 24b (317 mg, 89%) was synthesized from 23b in the same manner as 24a and was collected as a colorless oil. 1H-NMR (CDCl3, 300 MHz): δ 1.86 (quintet, J = 7.2 Hz, 2H), 2.13-2.20 (m, 2H), 2.46 (s, 3H), 3.00 (t, J = 7.2 Hz, 2H), 3.95 (s, 3H), 4.99-5.08 (m, 2H), 5.76-5.90 (m, 1H), 7.96 (s, 1H), 8.04 (s, 1H), 8.38 (s, 1H); 13C-NMR (CDCl3, 75.5 MHz): δ 21.3, 23.4, 33.2, 38.0, 52.4, 115.5, 126.7, 130.7, 132.8, 134.5, 137.5, 138.1, 139.0, 166.6, 199.7 MS (M+H)+ calcd: 247.1; found: 247.5.
tert-Butyl ((2R,3S)-3-(3-(hex-5-enoyl)-5-(N-methyl-methylsulfonamido)benzamido)-2-hydroxyoct-7-en-1-yl)(3-isopropylbenzyl)carbamate (25a). Aqueous LiOH (40 mg/mL, 324 µL, 0.54 mmol) was added to a solution of 24a (61 mg, 0.18mmol) in a mixture of dioxane (10 mL) and water (5 mL) and the solution was stirred at room temperature for 2 h. After neutrilization with 1 M HCl, the volatiles were evaporated and the crude remainder was dissolved in DMF (10 mL). DIPEA (105 µL, 0.60 mmol) and compound 19 (46 mg, 0.12 mmol) were added and the temperature was lowered to 0 °C. HATU (59 mg, 0.16 mmol) was added and the solution was allowed to obtain room temperature overnight. The DMF was subsequently evaporated and the crude product was purified using flash column chromatography (toluene/EtOAc 3:1 → toluene/EtOAc 1:1) to give 25a (73 mg, 89% over two steps) as a colorless glue. 1H-NMR (CDCl3, 300 MHz): δ 1.23 (d, J = 7.1 Hz, 6H), 1.47 (s, 9H), 1.50-1.67 (m, 4H), 1.78-1.91 (m, 2H), 2.00-2.20 (m, 5H), 2.80-2.93 (m, 1H), 2.87 (s, 3H), 3.02 (t, J = 7.3 Hz, 2H), 3.25-3.42 (m, 2H), 3.38 (s, 3H), 3.95 (br s, 2H), 4.23 (br s, 1H), 4.43 (br s, 2H), 4.87-5.09 (m, 4H), 5.67-5.89 (m, 2H), 6.94-7.29 (m, 3H), 7.34 (br s, 1H), 8.02-8.13 (m, 2H), 8.35 (br s, 1H); 13C-NMR (CDCl3, 75.5 MHz): δ 23.2, 24.1, 25.8, 28.5, 28.6, 29.3, 33.1, 33.6, 34.1, 35.7, 38.0, 50.2, 52.2, 54.0, 73.1, 81.3, 115.1, 115.6, 125.0, 125.1, 125.4, 125.8, 128.3, 128.7, 129.1, 136.0, 137.6, 137.9, 138.3, 138.4, 142.6, 149.5, 157.5, 166.0, 198.9 MS (2M+Na)+ calcd: 1417.7; found: 1417.1.
3-(Hex-5-enoyl)-N-((2R,3S)-2-hydroxy-1-((3-isopro-pylbenzyl)amino)oct-7-en-3-yl)-5-methylbenzamide (25b). Compound 25b (117 mg, 85% over two steps) was synthesized from 24b in the same manner as 25a using amine 18a instead of 19 and was collected as a white solid. [a]D +0.5 (c 0.1, MeOH). 1H-NMR (CD3OD, 300 MHz): δ 1.20 (d, J = 6.9 Hz, 6H), 1.34-1.73 (m, 6H), 1.74-1.86 (m, 2H), 1.87-2.01 (m, 1H), 2.04-2.20 (m, 3H), 2.47 (s, 3H), 2.79-2.93 (m, 1H), 2.96-3.18 (m, 3H), 3.83-3.92 (m, 1H), 3.95-4.05 (m, 1H), 4.14 (d, J = 13.2 Hz, 1H), 4.21 (d, J = 13.2 Hz, 1H), 4.88-5.08 (m, 4H), 5.73-5.92 (m, 2H), 7.21-7.38 (m, 3H), 7.73 (s, 1H), 7.96 (s, 1H), 8.19 (s, 1H); 13C-NMR (CD3OD, 75.5 MHz): δ 21.2, 24.2, 24.4, 26.6, 30.4, 34.1, 34.3, 35.1, 38.8, 51.0, 52.5, 54.3, 70.7, 115.2, 115.6, 128.4, 128.5, 128.6, 129.1, 130.1, 132.8, 133.3, 135.5, 138.6, 139.3, 139.5, 140.5, 151.2, 170.3, 201.9 HRMS calcd (M+H)+: 505.3430; found 505.3437. LC-MS Purity System A: tR = 3.15 min, 100%; System B: tR = 1.89 min, 100%.
tert-Butyl ((2R,3S)-3-(3-(hex-5-enoyl)-5-methylbenza-mido)-2-hydroxyoct-7-en-1-yl)(3-isopropylbenzyl)carba-mate (26). To a solution of 25b (40 mg, 0.079 mmol) in MeOH (8 mL) were added Boc2O (25 mg, 0.11 mmol) and TEA (22 µL, 0.16 mmol). The mixture was stirred at room temperature for 3 h, and the solvent was then evaporated. Flash column chromatography (toluene/EtOAc 6:1 → toluene/EtOAc 3:1) provided compound 26 (31 mg, 65%) as a colorless glue. 1H-NMR (CDCl3, 300 MHz): δ 1.22 (d, J = 6.9 Hz, 6H), 1.48 (s, 9H), 1.50-1.65 (m, 6H), 1.72-1.91 (m, 2H), 1.96-2.20 (m, 4H), 2.44 (s, 3H), 2.86 (septet, J = 6.9 Hz, 1H), 3.00 (t, J = 7.3 Hz, 2H), 3.29 (dd, J = 4.4, 14.6 Hz, 1H), 3.35-3.51 (m, 1H), 3.92 (br s, 1H), 4.03-4.27 (m, 2H), 4.33-4.56 (m, 2H), 4.87-5.09 (m, 4H), 5.67-5.90 (m, 2H), 6.92-7.28 (m, 3H), 7.78-7.90 (m, 2H), 8.18 (br s, 1H); 13C-NMR (CDCl3, 75.5 MHz): δ 21.4, 23.4, 24.1, 25.7, 28.5, 29.1, 33.2, 33.6, 34.2, 38.0, 50.4, 52.3, 53.7, 73.6, 81.2, 115.1, 115.5, 124.1, 125.0, 125.7, 125.8, 128.7, 131.6, 132.2, 134.8, 137.5, 137.7, 138.1, 138.4, 139.1, 149.5, 158.9, 167.2, 199.9 MS (M+H)+ calcd: 605.4; found: 604.9.
{(R)-2-Hydroxy-2-(E)-(S)-16-(methanesulfonyl-methyl- amino)-2,13-dioxo-3-aza-bicyclo[12.3.1]octadeca-1(17),8, 14,(18),15-tetraen-4-yl-ethyl}-(3-isopropyl-benzyl)-carba-mic acid tert-butyl ester (27a). Compound 25a (65 mg, 0.093 mmol) was dissolved in dry DCM (100 mL) and the solution was degassed with N2. Hoveyda-Grubbs catalyst 2nd generation (30 mg, 0.048 mmol) was added and the mixture was refluxed under N2 overnight. After evaporation of the solvent, the crude material was purified by flash column chromatography (toluene/EtOAc 3:1 → toluene/EtOAc 1:1) to yield 27a (13 mg, 21%) as a colorless glue. 1H-NMR (CDCl3, 300 MHz): δ 1.14-1.30 (m, 2H), 1.23 (d, J = 6.9 Hz, 6H), 1.34-2.38 (m, 10H), 1.42 (s, 9H), 2.65-3.15 (m, 3H), 2.87 (s, 3H), 3.20-3.53 (m, 2H), 3.35 (s, 3H), 3.93 (br s, 2H), 4.25-4.58 (m, 3H), 5.55-5.93 (m, 2H), 6.87-7.38 (m, 3H), 7.88-8.15 (m, 3H); 13C-NMR (CDCl3, 75.5 MHz): δ 24.8, 25.4, 27.3, 28.4, 29.0, 29.5, 31.2, 32.2, 36.4, 37.9, 38.3, 50.1, 52.5, 54.8, 74.1, 80.8, 124.2, 124.5, 125.1, 127.2, 128.5, 128.7, 129.9, 130.7, 133.1, 137.6, 137.5, 139.0, 143.1, 149.4, 158.0, 167.8, 199.5 MS (M+H)+ calcd: 670.4; found: 670.9.
(R)-2-Hydroxy-2-((E)-(S)-16-methyl-2,13-dioxo-3-aza- bicyclo[12.3.1]octadeca-1(17),8,14(18),15-tetraen-4-yl)-ethyl-(3-isopropyl-benzyl)-carbamic acid tert-butyl ester (27b). Compound 27b (14 mg, 47%) was synthesized in the same manner as 27a from 26 and was collected as a white powder. [a]D +16 (c 0.1, MeOH). 1H-NMR (CDCl3, 300 MHz): δ 1.21 (d, J = 6.9 Hz, 6H), 1.40 (s, 9H), 1.42-2.30 (m, 11H), 2.44 (s, 3H), 2.65-2.78 (m, 1H), 2.81-2.96 (m, 1H), 2.97-3.12 (m, 1H), 3.31 (dd, J = 4.1, 14.2 Hz, 1H), 3.35-3.52 (m, 1H), 3.84-4.07 (m, 2H), 4.34-4.62 (m, 3H), 5.71 (dt, J = 6.3, 15.4 Hz, 1H), 5.78-5.91 (m, 1H), 6.27 (br s, 1H), 6.97-7.28 (m, 3H), 7.78 (s, 1H), 7.86-7.96 (m, 2H); 13C-NMR (CDCl3, 75.5 MHz): δ 21.4, 24.2, 27.6, 27.7, 28.5, 29.8, 30.7, 30.9, 34.2, 37.7, 50.0, 52.5, 54.7, 73.9, 80.9, 125.0, 125.1, 125.6, 128.4, 128.7, 130.8, 131.5, 132.8, 133.3, 136.1, 136.3, 138.0, 140.2, 149.4, 157.9, 169.7, 200.9 HRMS calcd (M+H)+: 577.3641; found 577.3647. LC-MS Purity System A: tR = 3.70 min, 100%; System B: tR = 2.06 min, 98%.
N-{(E)-(S)-4-(R)-1-Hydroxy-2-(3-isopropyl-benzyla-mino)-ethyl-2,13-dioxo-3-aza-bicyclo[12.3.1]octadeca-1 (17),8,14(18),15-tetraen-16-yl}-N-methyl-methanesulfo-namide (28a). Compound 27a (13 mg, 0.019 mmol) was dissolved in a mixture of DCM (10 mL) and TFA (2.5 mL). Triethylsilane (TES) (10 µL, 0.063 mmol) was added and the solution was stirred at room temperature for 1 h. The volatiles were evaporated and the remainder was dissolved in DCM and washed with 1 M NaOH. The organic phase was dried, filtered and evaporated to yield 28a (10 mg, 91%) as a colorless glue. [a]D +7 (c 0.1, MeOH). 1H-NMR (CDCl3, 300 MHz): δ 1.24 (d, J = 7.1 Hz, 6H), 1.43-2.11 (m, 12H), 2.12-2.27 (m, 2H), 2.70-2.80 (m, 2H), 2.82-2.93 (m, 1H), 2.88 (s, 3H), 2.95-3.12 (m, 1H), 3.35 (s, 3H), 3.64-3.72 (m, 1H), 3.79 (s, 2H), 3.87-3.98 (m, 1H), 5.73 (dt, J = 6.3, 15.1 Hz, 1H), 5.80-5.91 (m, 1H), 6.08 (d, J = 8.8 Hz, 1H), 7.09-7.19 (m, 2H), 7.21-7.29 (m, 1H), 7.95-8.02 (m, 2H), 8.04-8.10 (m, 1H); 13C-NMR (CDCl3, 75.5 MHz): δ 24.2, 27.4, 28.4, 28.9, 29.8, 30.8, 34.2, 36.4, 37.6, 38.0, 51.3, 53.6, 54.2, 71.8, 125.4, 125.6, 125.7, 126.4, 127.2, 128.6, 130.0, 130.7, 133.0, 137.5, 138.0, 139.9, 143.5, 149.3, 168.0, 199.5 HRMS calcd (M+H)+: 570.3002; found 570.3011. LC-MS Purity System A: tR = 2.46 min, 97%; System B: tR = 1.57 min, 99%.
(E)-(S)-4-(R)-1-Hydroxy-2-(3-isopropyl-benzylamino) -ethyl-16-methyl-3-aza-bicyclo[12.3.1]octadeca-1(17),8, 14(18),15-tetraene-2,13-dione (28b). Compound 28b (10 mg, 91%) was synthesized from 27b in the same manner as 28a and was collected as a colorless glue. [a]D +11 (c 0.1, MeOH). 1H-NMR (CDCl3, 300 MHz): δ 1.24 (d, J = 6.9 Hz, 6H), 1.47-1.68 (m, 2H), 1.70-2.08 (m, 8H), 2.10-2.37 (m, 2H), 2.41 (s, 3H), 2.62-2.95 (m, 5H), 2.96-3.10 (m, 1H), 3.65 (q, J = 5.3 Hz, 1H), 3.76 (d, J = 13.5 Hz, 1H), 3.83 (d, J = 13.5 Hz, 1H), 3.86-4.00 (m, 1H), 5.67-5.91 (m, 2H), 5.98 (d, J = 8.5 Hz, 1H), 7.08-7.26 (m, 3H), 7.76 (s, 1H), 7.84-7.96 (m, 2H); 13C-NMR (CDCl3, 75.5 MHz): δ 21.4, 24.2, 27.5, 28.6, 29.3, 30.8, 30.9, 34.2, 37.8, 51.2, 53.7, 54.2, 72.2, 124.7, 125.2, 125.3, 125.7, 126.4 HRMS calcd (M+H)+: 477.3117; found 477.3118. LC-MS Purity System A: tR = 2.65 min, 96%; System B: tR = 1.76 min, 97%.
N-((S)-2-(Allyloxy)-1-((S)-2,2-dimethyl-1,3-dioxolan- 4-yl)ethyl)-3-(hex-5-enoyl)-5-methylbenzamide (29). Compound 24b (31 mg, 0.12 mmol) was dissolved in dioxane/H2O (2:0.5 mL) and aqueous LiOH (40 mg/mL, 150 ìL, 0.25 mmol) was added. The reaction mixture was stirred at room temperature for 6 h and then co-evaporated with toluene. The crude material was re-dissolved in DMF (1 mL) and (S)-2-(allyloxy)-1-((S)-2,2-dimethyl-1,3-dioxolan-4-yl)ethanamine (21 mg, 0.10 mmol), HATU (71 mg, 0.18 mmol) and DIPEA (65 ìL, 0.37 mmol) were added. The reaction mixture was stirred at room temperature for 4 h, evaporated and purified using flash column chromatography (toluene/EtOAc 3:1) to give compound 29 (25 mg, 57%) as a colorless oil. 1H-NMR (CDCl3, 400 MHz): δ 1.50 (s, 6H), 1.61-1.67 (m, 2H), 2.14-2.18 (m, 2H), 3.04 (s, 3H), 2.57 (t, J = 6.3 Hz, 2H), 3.59 (dd, J = 6.8, 11.6 Hz, 1H), 3.77 (dd, J = 6.0, 9.9 Hz, 1H), 3.88-3.92 (m, 2H), 3.99 (dd, J = 6.0, 9.9 Hz, 1H), 4.04 (d, J = 5.0 Hz, 2H), 4.56 (m, 1H), 4.98-5.00 (m, 1H), 5.01-5.03 (m, 1H), 5.12 (bs, 1H), 5.13-5.15 (m, 1H), 5.67-5.75 (m, 2H), 5.79-5.87 (m, 1H), 7.93 (s, 1H), 8.02 (s, 1H), 8.36 (s, 1H). MS (M+H)+ calcd: 416.2; found: 416.2.
N-((2S,3S)-1-(Allyloxy)-3,4-dihydroxybutan-2-yl)-3-(hex-5-enoyl)-5-methylbenzamide (30). Compound 30 (656 mg, 95%) was synthesized from 29 in the same manner as compound 3. 1H-NMR (CDCl3, 400 MHz): δ 0.95 (s, 1H), 1.54-1.60 (m, 2H), 2.13-2.18 (m, 2H), 2.36 (s, 3H), 2.53 (s, 1H), 2.74 (t, J = 6.3 Hz, 2H), 3.49 (dd, J = 4.7, 9.6 Hz, 1H), 3.53-3.57 (m, 2H), 3.70 (dd, J = 4.7, 9.7 Hz, 1H), 3.78 (dd, J = 1.3, 9.9 Hz, 1H), 3.99 (d, J = 4.9 Hz, 2H), 4.10-4.12 (m, 1H), 4.73 (s, 1H), 4.98-5.01 (m, 1H), 5.02-5.05 (m, 3H), 5.64-5.72 (m, 1H), 5.75.5.83 (m, 1H), 7.92 (s, 1H), 8.00 (s, 1H), 8.32 (s, 1H). MS (M+H)+ calcd: 376.2; found: 376.1.
(E)-(S)-4-((S)-1,2-Dihydroxy-ethyl)-16-methyl-6-oxa-3-aza bicyclo[12.3.1]octadeca-1(18),8,14,16-tetraene-2,13-dione (31). Compound 31 (533 mg, 88%) was synthesized from 30 in the same manner as compound 27a and was collected as a white powder. 1H-NMR (CDCl3, 400 MHz): δ 0.70 (s, 1H), 1.70-1.76 (m, 3H), 1.92-1.96 (m, 2H), 2.41 (s, 3H), 2.73 (t, J = 5.7 Hz, 2H), 3.48 (dd, J = 1.6, 9.8 Hz, 1H), 3.55 (dd, J = 4.0, 9.9 Hz, 1H), 3.70 (m, 1H), 3.73 (dd, J = 1.6, 9.8 Hz, 1H), 3.87 (dd, J = 4.0, 9.9 Hz, 1H), 4.02-4.06 (m, 2H), 4.11-4.13 (m, 1H), 5.54-5.63 (m, 2H), 6.16 (s, 1H), 7.63 (s, 1H), 7.79 (s, 1H), 7.92 (s, 1H). MS (M+H)+ calcd: 348.2; found: 348.1.
Toluene-4-sulfonic acid (S)-2-hydroxy-2-((E)-(S)-16-methyl-2,13-dioxo-6-oxa-3-aza-bicyclo[12.3.1]octadeca-1(18),8,14,16-tetraen-4-yl)-ethyl ester (32). Compound 32 (434 mg, 57%) was synthesized from 31 in the same manner as compound 4 and was collected as a white powder. 1H-NMR (CDCl3, 400 MHz): δ 1.36 (s, 1H), 1.71-1.76 (m, 2H), 1.92-1.96 (m, 2H), 2.37 (s, 3H), 2.39 (s, 3H), 2.73 (t, J = 5.7 Hz, 2H), 3.48 (dd, J = 3.1, 9.5 Hz, 1H), 3.58 (dd, J = 5.6, 9.9 Hz, 1H), 3.69-3.75 (m, 2H), 3.85 (dd, J = 5.7, 9.9 Hz, 1H), 4.00-4.07 (m, 2H), 4.26 (t, J = 5.6 Hz, 1H), 5.61-5.72 (m, 2H), 6.10 (s, 1H), 7.46 (d, J = 6.0 Hz, 2H), 7.72 (bs, 1H), 7.74 (bs, 1H), 7.89 (bs, 1H), 8.03 (d, J = 6.0 Hz, 2H). MS (M+H)+ calcd: 502.2; found: 502.1.
(E)-(S)-4-{(R)-2-1-(5-tert-Butyl-oxazol-2-yl)-1-methyl-ethylamino-1-hydroxy-ethyl}-16-methyl-6-oxa-3-aza-bicyclo[12.3.1]octadeca-1(18),8,14,16-tetraene-2,13-dione (33). Compound 32 (199 mg, 0.40 mmol) was dissolved in EtOH (1.5 mL). 2-(5-(tert-butyl)oxazol-2-yl)propan-2-amine (180 mg, 0.99 mmol) and DIEPA (1.5 mL) were added and the reaction mixture was refluxed for 4 days. The solvent was evaporated and the crude material was purified using prep. LC-MS to give compound 33 (11 mg, 5%). 1H-NMR (CDCl3, 400 MHz): δ 1.40 (s, 1H), 1.45 (s, 9H), 1.62 (s, 6H), 1.72-1.76 (m, 2H), 1.92-1.96 (m, 2H), 2.45 (s, 3H), 2.62 (dd, J = 5.7, 9.9 Hz, 1H), 2.73 (t, J = 4.3 Hz, 2H), 2.87 (dd, J = 5.7, 9.9 Hz, 1H), 3.48 (dd, J = 3.4, 9.4 Hz, 1H), 3.69-3.75 (m, 2H), 4.03-4.08 (m, 3H), 4.52 (s, 1H), 5.59-5.69 (m, 2H), 6.17 (s, 1H), 6.83 (s, 1H), 7.62 (bs, 1H), 7.79 (bs, 1H), 7.98 (bs, 1H). HRMS calcd (M+H)+: 512.3119; found 512.3109.
N-{(E)-(S)-4-(R)-1-Hydroxy-2-(3-isopropyl-benzyla-mino)-ethyl-2,13-dioxo-6-oxa-3-aza-bicyclo[12.3.1] octadeca-1(17),8,14(18),15-tetraen-16-yl}-N-methyl-methanesulfonamide (34). Compound 34 was synthesized according to the synthetic route for 28a, using compound 8 instead of 19, and was collected as a white solid. [a]D +41 (c 0.1, MeOH). 1H-NMR (CD3OD, 300 MHz): δ 1.23 (d, J = 6.9 Hz, 6H), 1.74-1.94 (m, 2H), 2.16-2.22 (m, 2H), 2.64-2.75 (m, 1H), 2.78-2.93 (m, 3H), 2.96 (s, 3H), 3.19-3.28 (m, 1H), 3.38 (s, 3H), 3.72 (dd, J = 3.9, 9.3 Hz, 1H), 3.83 (d, J = 6.9 Hz, 2H), 3.88-4.02 (m, 3H), 4.08-4.17 (m, 2H), 5.84 (dt, J = 5.1, 15.4 Hz, 1H), 5.98 (dt, J = 6.7, 15.4 Hz, 1H), 7.12-7.16 (m, 2H), 7.21-7.27 (m, 2H), 8.04-8.05 (m, 1H), 8.10-8.11 (m, 1H), 8.21-8.22 (m, 1H); 13C-NMR (CD3OD, 75.5 MHz): δ 22.8, 25.8, 30.0, 33.4, 34.5, 35.8, 36.6, 51.2, 52.7, 52.9, 68.3, 70.2, 124.8, 125.2, 125.9, 126.3, 126.9, 127.8, 128.3, 129.3, 131.9, 136.2, 136.4, 137.5, 142.6, 148.6, 167.2, 200.0. HRMS calcd (M+H)+: 572.2794; found 572.2797. LC-MS Purity System A: tR = 2.30 min, 100%; System B: tR = 1.69 min, 100%.
(E)-(S)-4-(R)-1-Hydroxy-2-(3-isopropyl-benzylamino)- ethyl-16-methyl-6-oxa-3-aza-bicyclo[12.3.1]octadeca-1 (17),8,14(18),15-tetraene-2,13-dione (35). Compound 35 was synthesized according to the synthetic route for 28a, using compound 24b instead of 24a and compound 8 instead of 19, and was collected as a white solid. [a]D +92 (c 0.1, MeOH). 1H-NMR (CDCl3, 300 MHz): δ 1.24 (d, J = 6.9 Hz, 6H), 1.77-1.98 (m, 2H), 2.11-2.31 (m, 2H), 2.48 (s, 3H), 2.74-2.82 (m, 2H), 2.84-2.92 (m, 2H), 2.94-3.04 (m, 1H), 3.59 (dd, J = 3.9, 9.0 Hz, 1H), 3.73-3.79 (m, 1H), 3.84 (d, J = 3.3 Hz, 2H), 3.90 (dd, J = 4.5, 12.6 Hz, 1H), 4.06-4.13 (m, 1H), 4.22 (dd, J = 2.7, 9.0 Hz, 1H), 4.25-4.31 (m, 1H), 5.80-5.98 (m, 2H), 7.09-7.14 (m, 2H), 7.19 (s, 1H), 7.22-7.23 (m, 1H), 7.91 (s, 1H), 7.98 (s, 1H), 8.00 (s, 1H); 13C-NMR (CDCl3, 75.5 MHz): δ 21.4, 24.1, 24.2, 26.9, 31.0, 34.2, 36.9, 51.1, 52.3, 54.1, 69.0, 69.6, 71.4, 124.0, 125.4, 125.8, 126.6, 128.6, 128.9, 131.9, 133.2, 134.0, 134.4, 136.2, 139.4, 140.3, 149.3, 166.7, 200.5 HRMS calcd (M+H)+: 479.2910; found 479.2925. LC-MS Purity System A: tR = 2.71 min, 98%; System B: tR = 1.67 min, 99%.
(E)-(S)-4-(R)-1-Hydroxy-2-(3-isopropyl-benzylamino)- ethyl-17-methyl-3-aza-bicyclo[13.3.1]nonadeca-1(18),9, 15(19),16-tetraene-2,14-dione (36). Compound 36 was synthesized according to the synthetic route for 28b, using compound 18b instead of 18a, and was collected as a white solid. [a]D +2.7 (c 0.1, MeOH). 1H-NMR (CD3OD, 300 MHz): δ 1.23 (d, J = 6.9 Hz, 6H), 1.29-1.50 (m, 2H), 1.56-1.74 (m, 4H), 1.78-1.89 (m, 2H), 2.02-2.23 (m, 4H), 2.44 (s, 3H), 2.54-2.64 (m, 1H), 2.77 (dd, J = 8.1, 12.6 Hz, 1H), 2.82-2.94 (m, 2H), 3.28-3.38 (m, overlapped of solvent peak, 1H), 3.71 (dt, J = 3.3, 7.8 Hz, 1H), 3.86 (d, J = 4.8 Hz, 2H), 3.88-3.95 (m, 1H), 5.43 (dt, J = 6.5, 15.2 Hz, 1H), 5.60 (dt, J = 6.9, 15.2 Hz, 1H), 7.15-7.19 (m, 2H), 7.23-7.28 (m, 2H), 7.70 (s, 1H), 7.90 (s, 1H), 8.06 (s, 1H); 13C-NMR (CD3OD, 75.5 MHz): δ 21.3, 24.4, 25.7, 27.8, 29.2, 31.2, 32.5, 32.8, 35.4, 38.2, 52.6, 54.2, 55.2, 73.3, 126.8, 127.0, 127.4, 128.0, 129.7, 130.8, 131.8, 133.4, 134.1, 137.2, 137.4, 138.8, 141.0, 150.6, 171.2, 202.8 HRMS calcd (M+H)+: 491.3274; found 491.3274. LC-MS Purity System A: tR = 2.96 min, 98%; System B: tR = 1.78 min, 100%.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
We gratefully thank the following personnel at Medivir AB: Kurt Benkestock for helping us with HRMS measurements, Elizabeth Hamelink and Cathrine Åhgren for providing the inhibition data and Per-Ola Johansson. Finally, we would like to acknowledge Medivir AB for performing the X-ray crystallography work and for financial support.
SUPPLEMENTARY MATERIALS
Supplementary material is available on the publishers web site along with the published article.
PATIENT’S CONSENT
Declared none.

