All published articles of this journal are available on ScienceDirect.

Considering Rotatability of Hydroxyl Groups for the Active Site Residues of MMP-13 in Retrospective Virtual Screening Campaigns
Abstract
Considering different orientation of hydroxyl and thiol groups of receptor residues such as Thr, Tyr, Ser and Cys is an option available on Glide docking software. This is an attempt that can provide more realistic ligand-receptor interactions. Matrix metalloproteinase 13 (MMP-13) is a suggested target for several diseases including osteoarthritis and cancer. MMP-13 was selected as a receptor with reported flexibility in the active site residues. Four residues in the MMP-13 active site were selected and their hydroxyl groups were made flexible during docking: Tyr241, Thr242, Tyr243 and Thr244. The ability of retrospective virtual screenings using a rigid receptor for discriminating between actives and decoys were compared to those using receptor with different combination of flexible residues. Statistical analysis of the results and inspecting the binding pose of the ligands suggested that the hydroxyl orientation of Tyr241, Thr242, Tyr243 and Thr244 (in particular Thr242 and to a lesser extent Thr244) had impacts on the MMP-13 docking results.
INTRODUCTION
MMP-13 belongs to a large family of metalloenzymes called matrix metalloproteinases [1]. MMP-13 was studied extensively for its suggested roles in some pathological conditions such as osteoarthritis and cancer [2, 3]. On the other hand the computational methods have been employed for study of MMP-13 interactions with small molecules. The availability of much structural information (several NMR and X-ray structures available for MMP-13 in PDB) made the computational approaches reasonable for finding the new selective inhibitors for MMP-13 [4, 5]. However this area has its own problems. One of the potential problems is the flexibility of the active site of MMP-13. Similar to some other MMP enzymes this flexibility could be either intrinsic or induced-fit. Induced fit mechanism of MMP-1 manifests itself by Arg 214 displacement after large ligand binding [6]. This type of flexibility allows the larger ligands to bind with the binding site with a modified conformation. A similar mechanism was also suggested for MMP-11 which has a Glu at that place [7]. MMP-3 the intrinsic flexibility was observed even in the presence of ligands with a same group at their P1 position. Analysis of different crystal structures of MMP-13 in the present study,are available as NMR structures [8] and a receptor-based 3D QSAR study results have suggested that there is flexibility in S1´ specificity loop of MMP-13 even after ligand binding [6].
The importance of the consideration of the flexibility in the virtual screening of MMPs enzymes has been suggested [8]. They are some approaches available for improving docking performance for specific targets including machine learning methods [9]. Different docking softwares deal with receptor flexibility differently. One of the approaches implemented in Glide (Schrödinger, LLC, New York, NY, 2011) is the ability to adopt different orientation for hydroxyl groups of residues Tyr, Thr and Ser. In this study we investigated the effects of introducing this flexibility for some residues in MMP-13 active site on retrospective virtual screenings outcome with various docking methods implemented in Glide.
METHODS
Structural Superimposition
The 35 halo x-ray structures of MMP-13 were retrieved form PDB. For metameric structures only the first model was kept. The water molecules were removed and all of the structures were superimposed to a reference structure. The RMSD histogram for all residues was graphed. The RMSD graph was employed for conducting flexible receptor docking. All of the above tasks were done in Chimera.
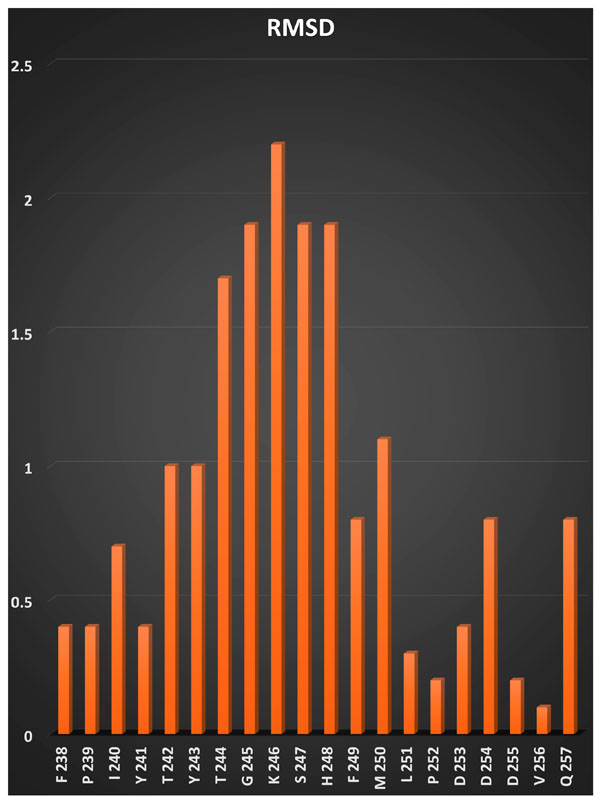
RMSD histogram for the active site residues of MMP-13.
Molecular Docking
The structure 3I7I was selected for the present study which according to a previous report is one of the best available MMP-13 3D structures for performing virtual screening. The Protein was prepared by Protein Preparation Wizard. In summary, the water molecules were removed, metal binding states were generated, H-bond assignment and restrained minimization were done. A grid box was centered on the center of the co-crystalized ligand and the ligands length was defined as 23 A°. Small molecules library consists of actives and decoys were prepared based on the DUD-E [10] collection for MMP-13 target. Duplicates were removed and 541 actives as well as 34521 decoys were docked in the MMP-13 active site by Glide. For most of the docking studies the HTVS (high-throughput virtual screening) setting or SP (standard precision) were employed except for the final round of screening in which the XP (extra precision) setting was employed. Due to limited computational resource, for XP docking 50 actives and 2500 decoys were randomly selected from the original DUD-E data set for MMP-13. After each retrospective virtual screening the results were assessed statistically by ROC curve analysis and enrichment factor. Glide can consider different orientations for hydroxyl group of residues Ser, Thr and Tyr. For Ser and Thr the hydroxyl group can orient in three minima while for Tyr it can adopted two orientations. We set the hydroxyl group of Tyr241, Thr242, Tyr243 and Thr244 rotatable with different docking precision.
Statistical Analysis
Using receiver operating characteristic (ROC) curve and enrichment factor (EF) for determining the discriminating power of a method was explained elsewhere [11]. In brief, the ROC curve and EF were applied to determine the performance of each conducted virtual screening. The increase in area under the curve (AUC) of ROC curve can be used as an indicator of improvement in discrimination between actives and decoys. EF is shown by the ability of a particular method to retrieve actives with a high rank among decoys and defined as below:
EF = (actives sampled / actives total) × (N total / N sampled)
All of the statistical test and plotting were done using R (R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL http://www.R-project.org/.) Including packages: enrichvs and ROCR [12].
RESULTS
Flexible Residues Identified by Structural Superimposition
RMSD of the residues in the active site of different experimental 3D structures for MMP-13 was shown in Fig. (1). As it was shown that the high variation in residues positon were observed in Ile240–Leu251. A previous study also reported the flexibility of Thr244-Gly250 residues (in particular Thr244) by investigating the two available NMR structures of MMP-13 [8]. The flexibility of Thr242 was also suggested previously [13]. According to the Glide available method for considering flexibility and accessibility of residues for ligands, the flexibility of hydroxyl groups of Thr242, Tyr243, Thr244 and Tyr245 were investigated.
The AUC of the ROC curve, EF2% and Ef1% obtained for each virtual screening run.
| Docking precision | Flexible part(s) | AUC of the ROC curve | EF10% | EF2% | EF1% |
|---|---|---|---|---|---|
| HTVS | Rigid | 0.641 | 3.09 | 5.89 | 8.08 |
| HTVS | Tyr241 | 0.616 | 2.89 | 6.07 | 8.35 |
| HTVS | Thr242 | 0.622 | 2.96 | 6.22 | 7.75 |
| HTVS | Tyr243 | 0.632 | 2.92 | 6.07 | 6.54 |
| HTVS | Thr244 | 0.644 | 2.90 | 6.10 | 8.02 |
| HTVS | Resides 241-244 | 0.643 | 2.92 | 6.02 | 8.76 |
| SP | Rigid | 0.660 | 3.40 | 7.79 | 9.38 |
| SP | Tyr241 | 0.657 | 3.30 | 8.07 | 10.46 |
| SP | Thr242 | 0.660 | 3.13 | 5.66 | 7.43 |
| SP | Tyr243 | 0.656 | 3.22 | 6.46 | 8.32 |
| SP | Thr244 | 0.662 | 3.34 | 6.90 | 9.73 |
| SP | Resides 241-244 | 0.654 | 3.24 | 7.09 | 10.28 |
| XP | Rigid | 0.724 | 4.60 | 15 | 20 |
| XP | Tyr241 | 0.722 | 4.20 | 11 | 20 |
| XP | Resides 241-244 | 0.722 | 4.20 | 15 | 24 |
Retrospective Virtual Screening
Table 1 summarizes the results of different virtual screening performed on MMP-13. For HTVS the EF1% for the virtual screening considered all four hydroxyl groups rotatable is higher than the one obtained for rigid docking. In docking with SP mode the EF1% of both docking which considered the hydroxyl groups of either Tyr241 or Tyr241-Thr244 rotatable are higher than EF1% of rigid docking. The difference between rigid and flexible docking is more obvious in SP docking compared to HTVS one. In XP docking the best performed flexible residues of previous runs were only selected. The results again showed an improvement in EF1% by considering the flexibility for all four residues.
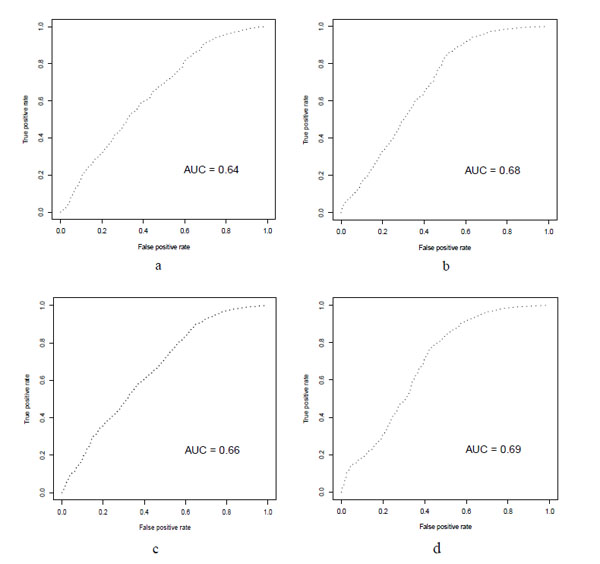
ROC curve for the Virtual screening results. a: HTVS docking with rigid receptor and Glide scoring. b: HTVS docking with rigid receptor and Glide lipo scoring. c: SP docking with rigid receptor and Glide scoring. d: SP docking with rigid receptor and Glide lipo scoring
Table 2 shows another finding of our study. Glide score takes into account a number of parameters. Glide lipo parameter is one of them that rewards favorable hydrophobic interactions. In all of the HTVS or SP virtual screenings the Gilde lipo scoring gave higher AUC accompanying with higher EF1% compared to Glide score. In the XP mode this was not seen that it could be due to the small sample size (~2500) were used for XP docking in comparison to HTVS and SP modes (~30000). The ROC curves were shown in Fig. (2).
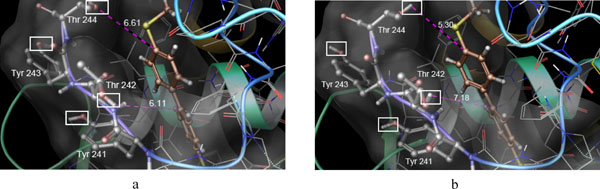
Binding pose of CHEMBL10335 in MMP-13 active site: after XP dock with rigid receptor (a) and XP dock with flexible receptor (b). The distances were shown in Angstrom. Rotatable hydroxyl groups are shown within white rectangles.
Inspection of the Binding Poses
By comparison of ranked ligand list it was found that some actives got better score in XP run with flexible residues compared to rigid XP run. As an example the active compound CHEMBL10335 was selected for furtherer examination. This was ranked 15 with docking score -9.861 in the rigid XP docking while ranked 3 with docking score of -12.172 in the flexible XP dock. As it is shown in Fig. (3) the hydroxyl groups of both Thr242 and Thr244 adopted different orientations in flexible docking. However, as the residue Thr242 was located near the hydrophobic part of the ligand it seems that the closer hydroxyl group to this hydrophobic part in rigid docking at least in part, was responsible for the higher energy of the docked pose for ligand CHEMBL10335. Furthermore, the hydroxyl group of the residue Tyr241 didn’t change upon binding of ligand CHEMBL10335 and this was seen in the binding of the most of the actives that rendered it as a lesser important residue in the bonding of the ligands.
DISCUSSION
Our results indicated that considering different orientation for hydroxyl group of of Tyr241, Thr242, Tyr243 and Thr244 in MMP-13 active site could slightly improve a retrospective virtual screening performance. The best performance was obtained by XP setting and considering the hydroxyl group rotatability for all of the four residues Tyr241-Thr244. By visual inspection of the docking poses the importance of flexibility of Thr242 was revealed. Furthermore, using the Glide lipo score improved the discriminating power of virtual screenings compared to standard glide score in our study.
Comparison of the obtained AUC of the ROC curve and Ef1% obtained by employing Glide score to those of Glide lipo score.
| Docking precision | Flexible part(s) | AUC of the ROC curve (docking score) | AUC of the ROC curve (Glide lipo) | EF1% (docking score) |
EF1% (Glide lipo) |
|---|---|---|---|---|---|
| HTVS | Rigid | 0.641 | 0.683 | 8.08 | 27.71 |
| HTVS | Resides 241-244 | 0.643 | 0.687 | 8.76 | 29.83 |
| SP | Rigid | 0.660 | 0.693 | 9.38 | 23.01 |
| SP | Resides 241-244 | 0.654 | 0.689 | 10.28 | 28.38 |
| XP | Rigid | 0.724 | 0.664 | 20 | 16 |
| XP | Resides 241-244 | 0.722 | 0.665 | 24 | 16 |
HTVS and SP docking modes of Glide are developed for screening of large database in a reasonable time. Thus, they are more forgiving than XP mode and considered some general flexibility for protein. XP method was designed to find ligands that can bind to the particular conformation of the given receptor. According to these facts the improvements in virtual screening results was more obvious in XP mode than two other methods. The improvements were in EF1% which is an important metric for assessment of a virtual screening performance. While the AUC of ROC curve indicates the average performance of a virtual screening on a particular subset the EF1% indicates the ability of a virtual screening in enriching the active compounds.
The MMP-13 active site is large, hydrophobic and flexible. The most of the zinc binding inhibitors of MMP-13 can only reaches to the earlier part of the active site (Tyr241-Thr244) while large ligands and those that classified as none zinc binding inhibitors [14] could bind deeply into part of the active site that consist of residues GLy245-Leu251. As it was shown in Fig. (1) and suggested by others [8] the flexibility of residues at the deep part of the MMP-13 active site is higher. However, as the most available inhibitors of MMP-13 (including those in DUD-E dataset) interacts with the shallower part of the MMP-active site. We investigated the considering the rotatability of hydroxyl groups of Tyr241-Thr244 on the virtual screening results. The study on flexibility of whole side chains of those in particular Thr242 could be a subject of another study. The better performance of Glide lipo score compared to Glide score may be related to the hydrophobic nature of the MMP-13 active site.
The present study provided a clue for considering flexibility of MMP-13 enzyme during virtual screening studies. As in this study only the flexibility of hydroxyl group of certain residues were considered and that showed improvement in virtual screening outcomes, considering the full flexibility of the side chains for different residues may have a greater influence on dockings and virtual screenings for MMP-13.
CONFLICT OF INTEREST
The author confirms that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
This work is supported financially by Mashhad University of Medical Sciences.
Patient’s Consent
Declared none.
REFERENCES
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]

