All published articles of this journal are available on ScienceDirect.

Discovery of Iminobenzimidazole Derivatives as Novel Cytotoxic Agents
Authors Info & Affiliations
Abstract
In our quest to identify inhibitors of the eukaryotic translation initiation factor 4F (eIF4F), we serendipitously discovered a novel cytotoxic agent. Even though this compound did not inhibit translation, we explored the structural requirements for its cytotoxicity due to its structural originality. A series of 1,3-disubstituted iminobenzimidazoles was synthesized and evaluated for their in vitro cytotoxicity. The structure-activity relationship studies demonstrate that hydrophobic substituent is essential for activity. The most active compounds displayed a cytotoxicity in KB, HL60 and HCT116 human cancer cells with an IC50 of about 1μM. These first-in-class series of low molecular weight synthetic molecules may provide the basis for the development of new anticancer drugs.
1. INTRODUCTION
Targeting the eukaryotic translation initiation factor 4F (eIF4F) holds promise as novel anticancer drugs that can overcome intra-tumor heterogeneity [1]. Indeed, the eIF4F complex, which regulates the cap-dependent protein synthesis is dysregulated in many types of cancers, leading to the overexpression of proteins that promote tumor growth, metastasis and resistance to cancer treatments. More important, several natural products that inhibit eIF4F, principally flavaglines, but also hippuristanol and pateamine, have demonstrated promising activities in several mouse models of cancers. In our quest to identify novel eIF4F inhibitors [2-6], we performed a virtual screening and we identified the iminobenzimidazole 1 (ChemBridge 5657657, Scheme 1). as a putative eIF4F inhibitor. Unfortunately, when we examine whether it inhibits eIF4F by using a bicistronic luciferase reporter construct [3], we didn’t find any significant activity. However, we found that it is cytotoxic in A375 melanoma cells. Considering that 1 had been described in a small number of patents and articles, none of them were related to cancer [7-10], and since the anticancer potential of iminobenzimidazoles has been scarcely investigated [11], we explored the requirement of 1 for its cytotoxicity to determine whether it could provide the basis for the development of new anticancer agents.
2. MATERIALS & METHODS
2.1. Chemical Syntheses
2.1.1. 2-[(4-Chlorophenoxy) Methyl] Oxirane (3)
4-Chlorophenol (12.0 g, 93.3 mmol, 1 equiv) was taken up into acetone (250 mL), then K2CO3 (38.7 g, 280.0 mmol, 3 equiv) and epichlorohydrin (21.6 mL, 280.0 mmol, 3 equiv) were added consecutively. The reaction mixture was set to stir at reflux for 24 h. At this time, an additional 3 equiv of epichlorohydrin was added and the solution was allowed to stir at reflux for an additional 24 h. The mixture was cooled down to room temperature, and the solids were filtered off. The solvent was removed under reduced pressure and the resulting oil was taken up in diethyl ether (150 mL). The organic layer was washed with H2O (1 x 150 mL), 1M aqueous NaOH solution (1 x 150 mL), and brine (1 x 150 mL). The organic layer was dried over MgSO4, filtered, and the solvent was removed under reduced pressure. The resulting yellow oil was purified via column SiO2 chromatography (10% EtOAc/hexanes) to yield the desired epoxide as a yellow oil (15.2 g, 88%); 1H NMR (CDCl3, 400MHz) δ 7.29-7.25 (m, 2H), 6.90-6.86 (m, 2H), 4.24 (dd, J= 2.8, 10.8 Hz, 1H), 3.94 (dd, J= 2.8, 10.8 Hz, 1H), 3.39-3.35 (m, 1H), 2.94 (t, J= 4.4 Hz, 1H), 2.78 (dd, J = 2.8, 4.4 Hz, 1H); 13C NMR (CDCl3, 100MHz) δ 157.1, 129.4, 126.1, 115.9, 69.1, 50.0, 44.6.
2.1.2. 1-(4-Chlorophenoxy)-3-(2-Imino-2,3-Dihydro-1H-Benzo[d]imidazol-1-yl)Propan-2-ol (4)
(4-Chlorophenoxy)-3-(2-imino-2,3-dihydro-1H-benzo[d]imidazol-1-yl)propan-2-ol (4). To a solution of 2-aminobenzimidazole 2 (4.0 g, 30.04 mmol) in H2O/Dioxane 35/35 mL, KOH (1.6 g, 30.04 mmol) and oxirane 3 (5.5 g, 30.04 mmol) were added. The mixture was stirred at 110 °C for 2 hours, then cooled to room temperature. Dioxane was removed under reduced pressure and the resulting solution was extracted 3 times with 50 mL of AcOEt. The organic layer was dried over MgSO4, filtered, and the solvent was removed under reduced pressure. The resulting solid was recrystallized with EtOH/H2O 3/7 to give the desired product as a white solid (6.0 g, 18.7 mmol, 63%). 1H NMR (DMSO, 400MHz) δ 7.34 (d, J= 8.4 Hz, 2H), 7.14 (t, J= 8.4 Hz, 2H), 6.90-6.94 (m, 3H), 6.84 ((t, J= 7.2 Hz, 2H), 6.26 (s, 2H), 5.59 (s, 1H), 3.93-4.15 (m, 5H). 13C NMR (DMSO, 100MHz) δ 157.8, 155.8, 143.1, 135.3, 129.7, 124.8, 120.7, 118.5, 116.7, 115.2, 108.3, 70.4, 68.3, 45.4.
2.1.3. 10-[3-(4-Chlorophenoxy)-2-Hydroxypropyl]-3,4-Dihydrobenzo [4,5] Imidazo [1,2-a] Pyrimidin-2(10H)-one (6)
To a solution of 4 (0.1 g, 0.31 mmol) in acetonitrile (4 mL), 3-bromo-N,N-dimethylpropanamide 5 (0.08 g, 0.43 mmol) was added and the mixture was stirred at reflux for 15 hours. K2CO3 (0.63 mmol) was added and the mixture was stirred at reflux for 2 days. Water (10 mL) and AcOEt (10mL) were added and the organic layer was extracted, dried over MgSO4 and concentrated. The residue was purified on flash chromatographic using (MeOH/DCM 5/95) to give the desired product as a white solid (0.7 mg, 55%). 1H NMR (400 MHz, DMSO) δ 7.48 (d, J = 7.6 Hz, 1H), 7.38 (d, J = 7.6 Hz, 1H), 7.32 (d J = 8.4 Hz, 2H), 7.15-7.23 (m, 2H), 6.95 (d, J = 8.4 Hz, 2H), 5.56 (d, J = 7.6 Hz, 2H), 4.25-4.31 (m, 1H), 4.12-4.21 (m, 4H), 3.93-4.03 (m, 2H), 2.57-2.61 (m, 2H). 13C NMR (400 MHz, DMSO) δ 175.3, 157.7, 155.4, 131.5, 130.0, 129.6, 124.8, 122.7, 122.5, 116.7, 110.7, 109.2, 70.8, 67.2, 45.3, 38.7, 29.8. LCMS-ESI (m/z) [M+H] + calcd: 372.10, found 372.02.
2.1.4. 9-[3-(4-Chlorophenoxy)-2-Hydroxypropyl]-3H-Benzo[d] Imidazo [1,2-a] Imidazol-2(9H)-One (8).
A mixture of 2 (0.1g, 0.31 mmol, 1 equiv) was taken up into 4 ml of acetonitrile, then bromoacetyl bromide 7 (0.02 mL, 0.31 mmol, 1equiv) and K2CO3 (0.08 g, 0.62 mmol, 2 equiv). The reaction mixture was stirred at reflux for 24 h, the mixture was cooled down to room temperature, and the solid was filtered, to yield the desired product as a white solid (0.126 g, 39%); 1H NMR (400 MHz, DMSO) δ 7.56 (d, J = 8.0 Hz, 1H), 7.17-7.35 (m, 5H), 6.955-6.980 (m, 2H), 5.55 (d, J = 2.4 Hz, 1H), 4.43 (s, 2H), 4.17-4.31 (m, 3H), 4.00-4.05 (m, 2H). 13C NMR (400 MHz, DMSO) δ 186.1, 168.5, 157.7, 133.7, 129.7, 129.2, 124.9, 123.4, 122.1, 116.7, 111.7, 109.7, 70.6, 67.1, 51.4, 46.9. LCMS-ESI (m/z) [M + H] + calcd: 358.09, found 358.08.
2.1.5. 1-(4-Chlorophenoxy)-3-(3-(2-Hydroxyethyl)-2-Imino-2,3-Dihydro-1H-Benzo[d] Imidazol-1-yl) Propan-2-ol (9b).
A mixture of 4 (0.1 g, 0.31 mmol, 1 equiv) was taken up into 4 ml of acetonitrile then 2-chloroethanol was added (0.04 mL, 0.62 mmol, 2.0 equiv). The reaction mixture was stirred at reflux for 24 h, the mixture was cooled down to room temperature. The solid was filtered, to yield the desired product as a white solid (0.04 g, 32%); 1H NMR (400 MHz, DMSO) δ 8.84 (bs, 1H), 7.59 (d, J= 9.2 Hz, 2H), 7.29-7.36 (m, 4H), 7.02 (d, J= 9.2 Hz, 2H), 5.65 (bs, 1H), 5.09 (bs, 1H), 4.24-4.34 (m, 5H), 4.06-4.09 (m, 2H), 3.74 (t, J= 4.8 Hz, 2H). 13C NMR (DMSO, 100MHz) δ 173.0, 157.7, 150.9, 130.8, 130.7, 129.7, 124.9, 123.6, 123.5, 116.8, 111.1, 70.3, 67.2, 58.9, 46.3, 45.8. LCMS-ESI (m/z) [M+H] + calcd: 362.12, found 362.05.
2.1.6. 2-[3-[3-(4-Chlorophenoxy)-2-Hydroxypropyl]-2-Imino-2,3-Dihydro-1H-Benzo[d] Imidazol-1-yl] Acetamide (9b)
A mixture of 4 (0.08 g, 0.25 mmol, 1 equiv) was taken up into 4 ml of acetonitrile, then chloroacetamide (0.02 mL, 0.25 mmol, 1.0 equiv) was added. The reaction mixture was stirred at reflux for 24 h, the mixture was cooled down to room temperature. The solid was filtered, to yield the desired product as a white solid (0.03 g, 31%); 1H NMR (400 MHz, DMSO) δ 8.96 (bs, 2H), 7.87 (s, 1H), 7.40-7.53 (m, 2H), 7.29-7.35 (m, 5H), 7.02 (d, J= 7.2 Hz, 2H), 4.90 (s, 2H), 4.29-4.36 (m, 3H), 4.07-4.09 (m, 2H). 13C NMR (400 MHz, DMSO) δ 167.1, 157.7, 151.3, 130.7, 130.5, 129.7, 124.9, 123.9, 123.8, 116.8, 111.3, 110.4, 70.3, 67.2, 46.4, 45.5. LCMS-ESI (m/z) [M+H] + calcd: 375.10, found 375.04.
2.1.7. [3-[2-[1, 3-Dioxolan-2-yl) Ethyl]-2-Imino-2, 3-Dihydro-1H-Benzo[d] Imidazol-1-yl]-3-(4-Chlorophenoxy) Propan-2-ol (9c)
A mixture of 4 (0.2 g, 0.63 mmol, 1 equiv) was taken up into 4 ml of acetonitrile, then 2-(2-bromoethyl-1,3 dioxane) (0.12 g, 0.69 mmol, 1.1 equiv) was added. The reaction mixture was stirred at reflux for 24 h, the mixture was cooled down to room temperature, and the solid was filtered and purified by column chromatography (AcOEt:MeOH / 9: 1), to yield the desired product as a brown solid (0.1 g, 38%); 1H NMR (400 MHz, DMSO) δ 7.59 (d, J = 7.6 Hz, 1H), 7.51 (d, J = 7.6 Hz,1H), 7.28-7.36 (m, 4H), 7.01 (d, J = 8.4 Hz, 2H), 4.91 (t, J = 4.2 Hz, 1H), 4.23-4.32 (m, 5H), 4.04-4.1 (m,2H), 3.73,3.88 (m,4H), 2.05 (q, J = 5.7 Hz, 2H) . 13C NMR (400 MHz, DMSO) δ 157.7, 150.4, 130.8, 129.9, 129.7, 125.0, 123.9, 123.8, LCMS-ESI (m/z) [M+H] + calcd: 418.12, found 418.14.
2.1.8. 1-(3-Allyl-2-Imino-2,3-Dihydro-1H-Benzo[d] Imidazol-1-yl)-3-(4-Chlorophenoxy)propan-2-ol (9d)
A mixture of 4 (0.12 g, 0.40 mmol, 1 equiv) was taken up into 4 ml of acetonitrile, then allylbromide (0.03 mL, 0.3 mmol, 1.0 equiv) was added. The reaction mixture was stirred at reflux for 24 h, the mixture was cooled down to room temperature, and the solid was filtered to yield the desired product as a white solid (0.05 g, 48%); 1H NMR (400 MHz, DMSO) δ 7.61 (dd, J= 7.8 Hz, J= 2.4 Hz, 1H), 7.52 (dd, J= 7.8 Hz, J= 2.4 Hz, 1H), 7.31-7.37 (m, 4H), 7.01 (d, J= 7.8 Hz, 2H), 5.83-5.91 (m, 1H), 5.38 (d, J= 7.6 Hz, 1H), 5.24 (d, J= 7.6 Hz, 1H), 4.82 (d, J= 3.2 Hz, 2H), 4.32-4.39 (m, 2H), 3.92-4.19 (m, 3H). LCMS-ESI (m/z) [M+H] + calcd: 357.12, found 357.13.
2.1.9. 1-(3-Benzyl-2-imino-2,3-Dihydro-1H-Benzo[d] Imidazol-1-yl)-3-(4-Chlorophenoxy)propan-2-ol (9e)
A mixture of 4 (0.09 g, 0.30 mmol, 1 equiv) was taken up into 4 ml of acetonitrile, then benzylbromide (0.05 g, 0.3 mmol, 1.0 equiv) was added. The reaction mixture was stirred at reflux for 24 h, the mixture was cooled down to room temperature, and the solid was filtered to yield the desired product as a white solid (0.06 g, 61%); 1H NMR (400 MHz, DMSO) δ 7.53 (d, J= 7.6 Hz, 1H), 7.18-7.37 (m, 10H), 7.01 (d, J= 7.6 Hz, 2H), 5.42 (s, 2H), 4.24-4.28 (m, 3H), 4.01-4.09 (m, 2H). 13C NMR (DMSO, 100MHz) δ 157.7, 150.8, 135.1, 130.8, 129.9, 129.7, 129.3, 128.4, 127.5, 124.9, 124.1, 124.0, 116.8, 111.6, 111.0, 70.4, 67.0, 46.6, 46.0. LCMS-ESI (m/z) [M+H] + calcd: 407.14, found 407.11.
2.1.10. 1-(4-Chlorophenoxy)-3-(2-Imino-3-(3-Phenylpropyl)-2,3-Dihydro-1H-Benzo [d] Imidazol-1-yl) Propan-2-ol (9f)
A mixture of 4 (0.09 g, 0.30 mmol, 1 equiv) was taken up into 4 ml of acetonitrile, then (3-bromopropyl)benzene (0.06 g, 0.3 mmol, 1.0 equiv) was added. The reaction mixture was stirred at reflux for 24 h, the mixture was cooled down to room temperature, and the solid was filtered to yield the desired product as a white solid (0.08 g, 65%); 1H NMR (400 MHz, DMSO) δ 7.15-7.28 (m, 10H), 7.02 (d, J= 7.6 Hz, 1H), 6.86 (d, J= 7.6 Hz, 2H), 4.35-4.71 (m, 5H), 4.04-4.14 (m, 2H), 2.89 (t, J= 7.6 Hz, 2H), 2.24 (t, J= 7.6 Hz, 2H). 13C NMR (DMSO, 100MHz) δ157.7, 150.3, 141.3, 130.7, 130.0, 129.7, 128.8, 128.5, 126.4, 124.9, 123.8, 123.8, 116.8, 111.4, 110.6, 70.4, 67.0, 46.4, 42.9, 32.3, 29.7. LCMS-ESI (m/z) [M+H] + calcd: 435.17, found 435.02.
2.1.11. 1-(4-Chlorophenoxy)-3-(3-(2-Cyclohexylethyl)-2-Imino-2,3-Dihydro-1H-Benzo[d] Imidazol-1-yl) Propan-2-ol (9g)
A mixture of 4 (0.2g, 0.63 mmol, 1 equiv) was taken up into 4 ml of acetonitrile, then 1-bromo-2-cyclohexyl ethane (0.132g, 0.69 mmol, 1.1 equiv) was added. The reaction mixture was stirred at reflux for 24 h, the mixture was cooled down to room temperature, and the solid was filtered, to yield the desired product as a white solid (0.126 g, 39%); 1H NMR (400 MHz, DMSO) δ 8.71 (sb, 1H), 7.6 (d, J = 8 Hz, 1H), 7.55 (d, J = 7.2 Hz, 1H), 7.3-7.36 (m, 4H), 6.99 (d, J = 8.8 Hz, 2H), 4.08-4.34 (m, 2H), 4.23-4.27 (m, 1H), 4.17 (t, J =7.8 Hz, 2H), 4.02-4.11 (m, 2H), 1.77 (d, J = 12.4 Hz, 2H), 1.63-1.69 (m,3H), 1.52-1.57 (q, J = 7.3 Hz, 2H), 1.34-1.38 (m,1H), 1.13-1.25 (m, 3H), 0.93-1.02 (m, 2H); 13C NMR (400 MHz, DMSO) δ 156.6, 148.1, 128.6, 127.6, 122.9, 121.8, 121.7, 114.7, 109.4, 108.5, 68.3, 64.9, 44.3, 39.2, 33.0, 32.9, 30.9, 24.3, 24.0 . LCMS-ESI (m/z) [M+H] + calcd: 428.12, found 428.18.
2.1.12. (E)-1-(4-Chlorophenoxy)-3-(2-Imino-3-(3-Phenylallyl)-2,3-Dihydro-1H-Benzo[d] Imidazol-1-yl)-Propan-2-ol (9h)
A mixture of 4 (0.2 g, 0.63 mmol, 1 equiv) was taken up into 4 ml of acetonitrile, then 3-bromo-1phenyl-1-propene (0.137g, 0.69 mmol, 1.1 equiv)) was added. The reaction mixture was stirred at reflux for 24 h, the mixture was cooled down to room temperature, and the solid was filtered, to yield the desired product as a white solid (0.186 g, 58%); 1H NMR (400 MHz, DMSO) δ 7.51 (t, J = 3.2, Hz,2H), 7.42 (d, 7.2 Hz, 2H), 7.25-7.35 (m, 7H), 7.0 (d, J = 8.8 Hz, 2H), 6.7 (d, J = 16Hz, 1H), 6.39 (dt, J = 15.6, 5.7 Hz, 1H), 5.0 (d, J = 5.2, 2H), 4.27-4.33 (m, 3H), 4.04-4.12 (m,2H), 13C NMR (400 MHz, DMSO) δ 157.8, 150.7, 136.2, 136.1, 133.1, 131.7, 130.9, 130.1, 129.8, 129.2, 128.6, 126.9, 124.9, 123.8, 122.6, 116.8, 111.2, 110.7, 70.5, 67.1, 46.4, 44.6. LCMS-ESI (m/z) [M+H] + calcd: 434.1, found 434.1.
2.1.13. 1-(4-Chlorophenoxy)-3-(2-Imino-3-(3-Phenylprop-2-yn-1-yl)-2,3-Dihydro-1H-Benzo [d] Imidazol-1-yl) Propan-2-ol (9i)
A mixture of 4 (0.15 g, 0.47mmol, 1 equiv) was taken up into 4 ml of acetonitrile, then 3-chloro-1phenyl-1-propyne (0.077 g, 0.51 mmol, 1.1 equiv) was added. The reaction mixture was stirred at reflux for 24 h and cooled down to room temperature. The solid was filtered to yield the desired product as a white solid (0.143 g, 66%); 1H NMR (400 MHz, DMSO) δ 7.7 (d, J = 7.6 Hz, 1H), 7.62 (d, J = 8, 1H), 7.31-7.44 (m, 9H), 7.0 (d, J = 9.6, 2H), 5.5 (s, 2H), 4.33-4.42 (m,2H), 4.23-4.27 (m,1H), 4.06-4.1 (m, 2H), 13C NMR (400 MHz, DMSO) δ 157.7, 150.3, 132.1, 130.9, 129.8, 129.7, 129.5, 129.3, 124.9, 124.2, 124.1, 121.6, 116.9, 116.8, 111.6, 110.9, 85.3, 82.6, 70.4, 67.1, 46.6, 33.9 . LCMS-ESI (m/z) [M+H] + calcd: 432.1, found 432.1.
2.1.14. 1-(4-Chlorophenoxy)-3-(2-Imino-3-(2-Phenoxyethyl)-2,3-Dihydro-1H-Benzo[d] Imidazol-1-yl) Propan-2-ol (9j)
A mixture of 4 (0.2 g, 0.63 mmol, 1 equiv) was taken up into 4 ml of acetonitrile, then 2-phenoxyethyl bromide (0.138 g, 0.69 mmol, 1.1 equiv) was added. The reaction mixture was stirred at reflux for 24 h and cooled down to room temperature. The solid was filtered to yield the desired product as a white solid (0.27 g, 85%); 1H NMR (400 MHz, DMSO) δ 7.63 (d, J = 7.6 Hz, 1H), 7.55 (d, J= 7.6 Hz, 1H), 7.23-7.33 (m, 6H), 6.99 (d, J = 8.8, 2H), 6.92 (t, J = 7.4 Hz, 1H), 6.82 (d, J = 8 Hz, 2H), 4.6 (m, 2H), 4.23-4.32 (m, 5H), 4.02-4.1 (m, 2H), 13C NMR (400 MHz, DMSO) δ 158.3, 157.7, 151.1, 130.8, 130.6, 130.2, 130, 129.7, 124.9, 123.6, 121.5, 116.8, 114.8, 111.2, 111.1, 111, 70.4, 67.1, 65.6, 46.4, 42.9. LCMS-ESI (m/z) [M+H] + calcd: 438.11, found 438.21.
2.1.15. 1-(3-(4-Bromophenethyl)-2-Imino-2,3-Dihydro-1H-Benzo[d] Imidazol-1-yl)-3-(4-Chlorophenoxy) Propan-2-ol (9k)
A mixture of 4 (0.2 g, 0.63 mmol, 1 equiv) was taken up into 4 ml of acetonitrile, then 1-bromoethyl – 4-(2-bromoethylbenzene) (0.18 g, 0.69 mmol, 1.1 equiv) was added. The reaction mixture was stirred at reflux for 24 h and cooled down to room temperature. The solid was filtered to yield the desired product as a white solid (0.144 g, 46%); 1H NMR (400 MHz, DMSO) δ 7.53-7.55 (m, 1H), 7.47 (d, J =7.6 Hz, 3H), 7.35 (d, J = 8.8 Hz, 2H), 7.27 (d, J =7.6 Hz, 4H), 7.0 (d, J = 8.8 Hz, 2H), 4.38 (t, J = 7.4, 2H), 4.21-4.29 (m, 3H), 4.02-4.08 (m, 2H), 4.97 (t, J = 7.4 Hz, 2H) . 13C NMR (400 MHz, DMSO) 157.7, 152.6, 150.6, 137.2, 131.9, 131.6, 130.7, 129.9, 129.8, 124.9, 123.6, 120.4, 116.8, 111.0, 110.5, 70.4, 67.2, 46.2, 43.7, 31.9. LCMS-ESI (m/z) [M+H] + calcd: 499.1, found 499.9.
2.1.16. 1-(4-Chlorophenoxy)-3-[2-Imino-3-[2-(Naphthalen-1-yl) Ethyl]-2, 3-Dihydro-1H-Benzo[d] Imidazol-1-yl] Propan-2-ol (9l)
A mixture of 4 (0.2g, 0.63 mmol, 1 equiv) was taken up into 4 ml of acetonitrile, then 1-(2-bromoethyl) naphthalene (0.16 g, 0.69 mmol, 1.1 equiv) was added. The reaction mixture was stirred at reflux for 24 h and cooled down to room temperature. The solid was filtered to yield the desired product as a white solid (0.281 g, 88%); 1H NMR (400 MHz, DMSO) δ 8.08 (s, 2H), 8.06 (d, J = 8.4 Hz, 1H), 7.9 (d, J = 7.6 Hz, 1H), 7.77-7.79 (m, 1H), 7.5-7.58 (m, 3H), 7.35-7.38 (m, 4H), 7.21(t, J = 7.4 Hz, 1H), 7.08-7.132 (m, 2H), 7.00-7.06 (m, 2H), 5.63 (s, 1H), 4.59 (t, J = 6.6 Hz, 2H), 4.23-4.31 (m, 3H), 4.00-4.09 (m, 2H), 3.5 (t, J = 6.8 Hz, 2H). 13C NMR (400 MHz, DMSO) δ 157.7, 150.3, 133.9, 133.8, 131.9, 130.4, 129.9, 129.8, 129.1, 127.8, 127.5, 126.7, 126.2, 125.9, 125.0, 123.7, 116.8, 111.3, 110.4, 70.4, 67.1, 46.3, 43.7, 30.2. LCMS-ESI (m/z) [M+H] + calcd: 472.2, found 472.1.
2.1.17. 1-(4-Chlorophenoxy)-3-(3-(3,3-Diphenylpropyl)-2-Imino-2,3-Dihydro-1H-Benzo [d] Imidazol-1-yl) Propan-2-ol (9m)
A mixture of 4 (0.2g, 0.63 mmol, 1 equiv) was taken up into 4 ml of acetonitrile, then 3,3-diphenyl-1-bromopropane (0.18 g, 0.69 mmol, 1.1 equiv) was added. The reaction mixture was stirred at reflux for 24 h and cooled down to room temperature. The solid was filtered to yield the desired product as a white solid (0.37 g, 86%); 1H NMR (400 MHz, DMSO) δ 8.74 (s, 2H), 7.52-7.55 (m, 1H), 7.26-7.35 (m, 12H), 7.18 (t, J = 6.8 Hz, 2H), 7.10-7.16 (m, 1H), 7.01 (d, J = 8.8 Hz, 2H), 5.59 (s, 1H), 4.21 (s, 3H), 4.02-4.14 (m, 5H), 5.53-5.55 (m, 2H). 13C NMR (400 MHz, DMSO) δ 157.7, 150.1, 144.4, 144.3, 130.6, 129.7, 129.0, 128.0, 127.8, 126.8, 124.9, 123.8, 116.8, 111.4, 110.2, 70.4, 67.0, 48.4, 46.3, 42.1, 32.7. LCMS-ESI (m/z) [M+H] + calcd: 512.2, found 512.11.
2.1.18. N-Methyl-3-Nitropyridin-2-Amine (11)
To a solution of methylamine (10 mL, 80.7 mmol) at 0°C, 2-chloro-3-nitropyridine 10 (1.6 g, 10.0 mmol) was added in several portions. The solution was stirred 1 hour at 0°C then 2 hours at room temperature before concentration in vacuum. The residue was washed with water (20 mL) and then extracted 3 times in AcOEt (20 mL). The organic layer was dried over MgSO4 then concentrated to give the desired product as a yellow oil (1.3 g, 85%). 1H NMR (400 MHz, CDCl3) δ 8.40-8.45 (m, 2H), 8.24 (bs, 1H), 6.62-6.65 (m, 1H), 3.16 (d, J = 2.4 Hz, 3H). 13C NMR (400 MHz, CDCl3) δ 155.7, 153.2, 135.2, 128.3, 111.5, 28.1.
2.1.19. N-Methylpyridine-2,3-Diamine (12)
To a solution of 11 (1.1 g, 7.18 mmol) in dry MeOH (25 mL), palladium 10% carbon (0.07 g, 0.71 mmol) was added. The mixture was stirred at room temperature and under hydrogen (atmospheric pressure) overnight. The solution was filtered over celite then concentrated to give the desired product as a yellow oil (0.88 g, 99%). 1H NMR (400 MHz, CDCl3) δ 7.78 (d, J = 5.2 Hz, 1H), 6.83 (d, J = 5.2 Hz, 1H), 6.51 (t, J = 5.4 Hz, 1H). 4.20 (bs, 1H), 3.18 (bs, 2H), 3.00 (d, J = 2.4 Hz, 3H). 13C NMR (400 MHz, CDCl3) δ 151.1, 139.2, 128.5, 121.7, 113.2, 28.7.
2.1.20. 3-Methyl-3H-Imidazo[4,5-b] Pyridin-2-Amine (13)
To solution of 12 (0.6 g, 4.87 mmol) in MeOH/H2O 12/12 mL, cyanogen bromide (1.5 g, 14.63 mmol) was added. The mixture was stirred at 60°C for 4 hours. After cooling to room temperature, MeOH was evaporated and the solution was basified to pH 8 using a solution of NaOH 1N. The organic layer was extracted 3 times with AcOEt (20 mL), dried over MgSO4 and concentrated in vacuum. The residue was purified by flash chromatographic (MeOH/DCM 1/9) to give the desired product as a brown solid (0.37 g, 55%). 1H NMR (400 MHz, DMSO) δ 7.84 (d, J = 7.2 Hz, 1H), 7.39 (dd, J = 7.2 Hz, J = 1.8 Hz, 1H), 6.92-6.96 (m, 1H), 6.78 (s, 2H). 3.51 (s, 3H). 13C NMR (400 MHz, DMSO) δ 156.5, 148.5, 137.8, 136.3, 120.5, 117.1, 27.3.
2.1.21. 2.1.21 1-(4-Chlorophenoxy)-3-(2-Imino-3-Methyl-2,3-Dihydro-1H-Imidazo[4,5-b] Pyridin-1-yl) Propan-2-ol (14)
To a solution of 3-methyl-3H-imidazo [4,5-b]pyridin-2-amine (0.1 g, 0.67 mmol) in H2O/dioxane 3/3 mL, KOH (0.04 g, 0.67 mmol) and epoxide 3 (0.13 g, 0.74 mmol) were added. The mixture was stirred at 110 °C for 2 hours, then cooled down to room temperature. Dioxane was removed under reduced pressure and the resulting solution was extracted 3 times with 50 mL of AcOEt. The organic layer was dried over MgSO4, filtered, and the solvent was removed under reduced pressure. The resulting solid was recrystallized with EtOH/H2O 3/7 to give the desired product as a white solid (0.05 g, 0.16 mmol, 25%). 1H NMR (CDCl3, 400MHz) δ 8.11 (d, J = 4.4 Hz, 1H), 7.65 (d, J = 7.6 Hz, 1H), 7.28 (d, J = 7.6 Hz, 1H), 6.87 (d, J = 7.6 Hz, 1H), 5.08 (bs, 1H), 4.31-4.42 (m, 1H), 3.94-4.09 (m, 3H), 3.76-3.80 (m, 1H), 3.64 (s, 3H). 13C NMR (400 MHz, CDCl3) δ 157.1, 155.4, 147.9, 139.7, 134.2, 129.4, 126.1, 122.4, 117.7, 115.7, 70.1, 69.4, 46.5, 26.8. LCMS-ESI (m/z) [M+H] + calcd: 333.10, found 333.04.
2.2. Biological Assays
2.2.1. Cell Culture
The human cell lines KB (nasopharyngeal epidermis carcinoma) and HCT116 (colon adenocarcinoma) were purchased from ECACC (Salisbury, UK) and HL60 (promyeocytic leukaemia) cells from ATCC. KB cells were grown in D-MEM medium supplemented with 10% fetal calf serum, in the presence of penicillin, streptomycin and fungizone in a 75 cm2 flask under 5% CO2, whereas the two other cell lines were grown in complete RPMI medium.
2.2.2. Cell Cytotoxicity Assay
Cells were plated in 96-well tissue culture plates in 200 μl medium and treated 24 h later with complexes dissolved in DMSO at concentrations ranged 0.5 nM to 10 μM and were prepared by using a Biomek 3000 (Beckman-Coulter). Control cells received the same volume of DMSO (1% final volume). After 72 h exposure to the drug, Cell Titer 96®AQuerous one reagent (Promega) was added and incubated for 3h at 37 °C: the absorbance was monitored at 490 nm and results are expressed as the inhibition of cell proliferation and cell viability calculated as the ratio [(1-(OD490 treated/OD490 control)) ×100]. For IC50 determinations (50% inhibition of cell viability), experiments were performed in duplicate.
3. RESULTS AND DISCUSSION
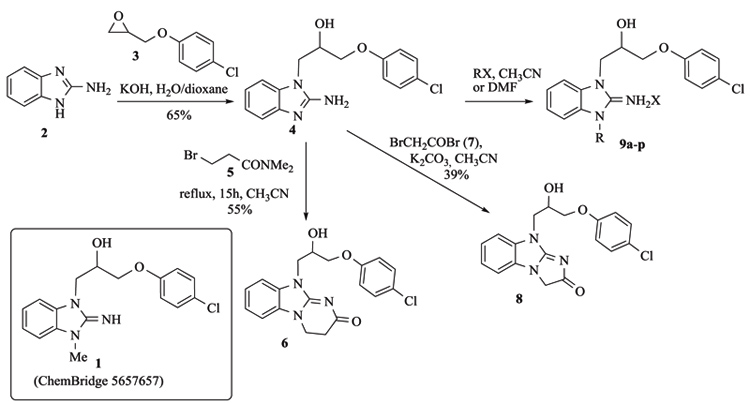
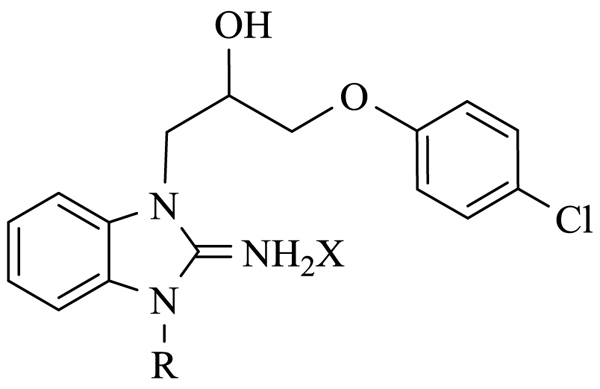
We started our Structure-Activity Relationships (SAR) investigation by replacing the N-methyl of 1 by various substituents. The synthesis of these derivatives began with the alkylation of aminobenzimidazole 2 with an epoxide 3 to afford adduct 4 Scheme (1). Condensation of 4 with acyl halides 5 and 7 delivered the cyclized adducts 6 and 8. Reaction with alkyl halides afforded adducts 9a-p (Scheme 1).
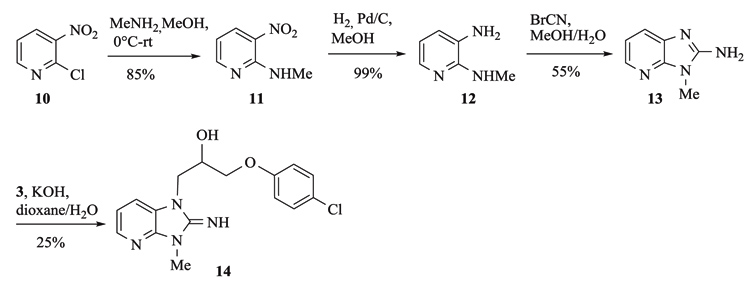
Aza-isostere 14 was prepared in 4 steps starting from 2-chloro-3-nitropyridine 10 (Scheme 2). Condensation with methylamine, followed by a reduction of the nitro group and ring closure with cyanogen bromide afforded the aza-benzimidazole 13 that was alkylated by epoxide 3 to yield 14.
Surprisingly, neither 1 nor its desmethyl analogue 4 was cytotoxic to KB cells at 10 μM (Table 1). The inclusion of the amidine moiety in an extra cycle (6, 8) or introduction of polar functions (9a-c) did not promote cytotoxicity. Introduction of a nitrogen in the benzimidazole moiety (14) was also ineffective, but the replacement of the methyl by an allyl (9d) or benzyl (9e) made these compounds extremely cytotoxic. Gratefully, increasing the size of the linker between iminobenzimidazolium and phenyl from one to three methylenes (9f) enhanced cytotoxicity.
Aza-isostere 14 was prepared in 4 steps starting from 2-chloro-3-nitropyridine 10 Scheme. (2). Condensation with methylamine, followed by a reduction of the nitro group and ring closure with cyanogen bromide afforded the aza-benzimidazole 13 that was alkylated by epoxide 3 to yield 14. Synthesis of aza-isostere 14.
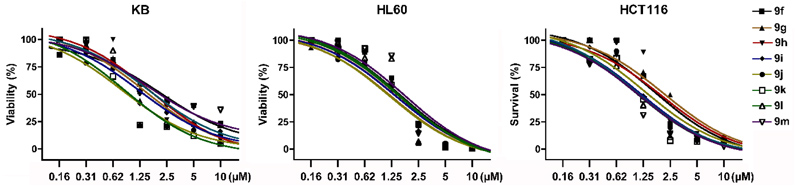
With the identification of potent cytotoxic compounds, we pursued our investigation by determining the IC50 of newly synthesized compounds in 3 human cancer cell lines (Table 2), (Fig. 1). These three human cancer cell lines are respectively derived from patients with cervical carcinoma, acute promyelocytic leukemia and colorectal carcinoma. As such, they represent a variety of different cancers. Replacement of the phenyl by a cyclohexyl (9g), introduction of unsaturations or an ether in the linker (9h-j), and the addition of a bromine (9k) or extra aromatic cycles (9l,m) did not significantly modified cytotoxicity, which was very similar in the 3 examined cell lines. These results suggest that these compounds probably bind with a comparable affinity for their target and indicate the requirement for a hydrophobic moiety connected to the benzimidazole ring by a linker with 2 or 3 carbons (Fig. 2).
| Cpd. | R | R’ | A | HX | 10-5M | 10-6M |
|---|---|---|---|---|---|---|
| 1 | Me | H | CH | - | 0±17 | 0±2 |
| 4 | H | H | CH | - | 0±9 | 0±3 |
| 6 | 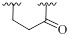 |
CH | - | 0±3 | 0±2 | |
| 8 | 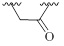 |
CH | - | 0±6 | 0±7 | |
| 9a | 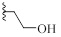 |
H | CH | HCl | 0±12 | 0±15 |
| 9b | 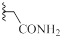 |
H | CH | HCl | 0±2 | 0±9 |
| 9c | 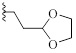 |
H | CH | - | 1±11 | 0±13 |
| 14 | Me | H | N | - | 0±7 | 0±4 |
| 9d | 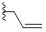 |
H | CH | HBr | 88±5 | 0±17 |
| 9e |  |
H | CH | HBr | 96±2 | 8±8 |
| 9f |  |
H | CH | HBr | 98±1 | 57±5 |
| PD | R | HX | KB | HL60 | HCT116 |
|---|---|---|---|---|---|
| 9f | 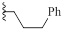 |
HBr | 2/1.2 | 1.1/1.0 | 1.5/1.5 |
| 9g | 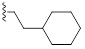 |
HBr | 1.3/1.5 | 1.25/1.25 | 1.3/2 |
| 9h | 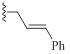 |
HBr | 2/1.2 | 1.25/1.25 | 2/2 |
| 9i | 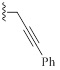 |
HBr | 1.75/0.8 | 1.75/1.5 | 1.3/2 |
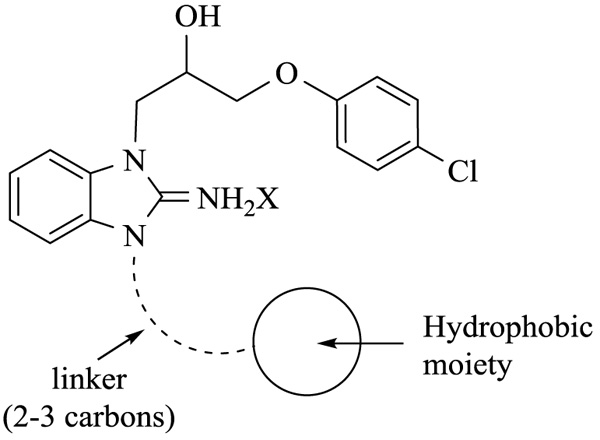
CONCLUSION
The research and development of a novel type of drugs are of great importance to improve the treatment of cancers. In this context, we describe herein the discovery and optimization of the first-in-class series of cytotoxic agents. The SAR studies demonstrated the requirement for a hydrophobic moiety. These results are highly encouraging, and SAR studies to examine structural variations of the iminobenzimidazole and 3-(4-chlorophenoxy)-2-hydroxypropyl moieties are underway.
CONSENT FOR PUBLICATION
Not applicable.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
This work is published within the LABEX ANR-10-LABX-0034_Medalis and received financial support from the French government managed by ANR under “Programme d’investissement d’avenir”. NC received a fellowship from Frères Mentouri University-Constantine 1.

