All published articles of this journal are available on ScienceDirect.

Hexacyclododecylamines with Sigma-1 Receptor Affinity and Calcium Channel Modulating Ability
Authors Info & Affiliations
Abstract
Introduction:
Recent research points to the Sigma Receptor (σR) as a possible neuromodulatory system with multi-functional action and σ1Rs have been suggested as a drug target for a number of CNS conditions. Hexacyclododecylamines have shown σ1R activity and provide an advantageous scaffold for drug design that can improve the blood-brain barrier permeability of privileged structures.
Methods and Materials:
A series of oxa- and aza- hexaxcyclododecylamines were synthesised and evaluated for sigma-1 receptor activity and voltage-gated calcium channel blocking ability to determine the effect of inclusion of amine containing heterocycles.
Results & Discussion:
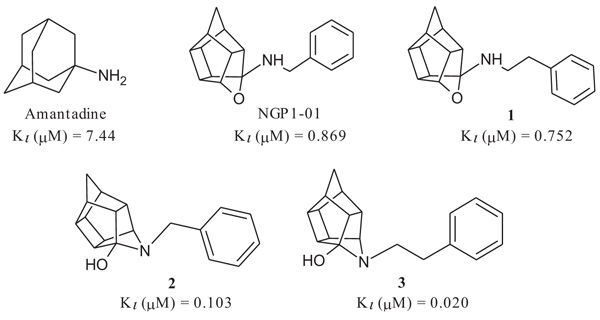
The compounds had promising σ1R activities (Ki = 0.067 – 11.86 µM) with the aza-hexacyclododecylamines 12, 24 and 27 showing some of the highest affinities (Ki = 0.067 µM, 0.215 µM and 0.496 µM respectively). This confirms, as observed in previous studies, that the aza compounds are more favourable for σ1R binding than their oxa counterparts. The addition of the amine heterocycle showed affinities similar to that of related structures with only two lipophilic binding regions. This indicates that the inclusion of an amine heterocycle into these structures is a viable option in the design of new σ1R ligands. Significant voltage-gated calcium channel blocking ability was also observed for 12, 24 and 27, suggesting a link between σ1R activity and intracellular calcium levels.
Conclusion:
The σ1R activity and potential effect on other receptor classes and calcium channels could prove beneficial in pharmacological application.
1. INTRODUCTION
The sigma receptor, which was originally thought to be an opioid receptor, is divided into two subtypes; the Sigma-1 Receptor (σ1R) and the Sigma-2 Receptor (σ2R) which are now classified as distinct receptors [1, 2] and do not share homology with any other known mammalian enzyme or receptor [3-5]. The σ1R has been cloned and is implicated in intracellular signalling, synaptic transmission, apoptosis and is able to mediate effects on calcium conductance through Voltage-Gated Calcium Channels (VGCC), N-methyl-D-Aspartate (NMDA) receptor activity, potassium channel activity, protein kinases and modulation of inositol phosphatases [6-8]. The σ1R receptor is located on the endoplasmic reticulum and it is likely that the σ1R mediates its response via translocation from the endoplasmic reticulum to other cellular compartments [9, 10].
The role of sigma receptors in mediating neuroprotection or neurodegeneration has been studied in several in vitro models of Central Nervous System (CNS) injury [11-13]. A consistent positive correlation between sigma neuroprotective potency and σ1R binding site affinity has been demonstrated, suggesting that the functional neuroprotective effect of σR ligands may be mediated by binding at the σ1R site [11]. Activation of σ1R receptors results in a complex, bipolar modulation of calcium homeostasis. It facilitates the mobilisation of inositol triphosphate receptor-gated intracellular calcium pools at the endoplasmic reticulum level and modulates extracellular calcium influx through VGCC at the plasma membrane level [12, 13]. Overstimulation of σ1 receptors may contribute to toxic intracellular calcium concentrations and sigma receptors are thus a target for neuroprotective drugs aimed at calcium modulation [12, 13]. Additionally, the σ1R receptors and the ER play an important functional role with the mitochondria, where the ER-to-mitochondria miscommunication has been implicated in calcium exchange and other cellular processes like mitophagy [14].
Since the discovery of σRs,much research was done in this field due to the implication of these receptors in various major (CNS) diseases [15, 16]. Some of the earliest σR ligands identified were diverse clinical antipsychotics that bind to σ1Rs with nanomolar affinity [17] and several antidepressants from unrelated pharmacological classes that interact with σ1Rs with high affinity [18, 19]. The role of σ1R in Alzheimer’s disease [20], Parkinson’s disease [20], anxiety disorders [21], depression [22] and drug addiction [23] is well accepted.
Polycyclic amines, such as the hexacyclododecylamine (HCD; also described as pentacycloundecylamines in literature) and the adamantane amines, were shown to be valuable scaffolds in the development of CNS drugs [24, 25]. These polycyclic compounds possess their own neuromodulatory activity on important receptor classes that have been implicated in CNS disease states such as Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, schizophrenia, stroke and disease states as intricate as drug addiction [24-26]. They act as NMDA receptor antagonists [27, 28], are able to spontaneously increase dopamine release [29] and have been shown to have neuroprotective activity through modulation of VGCC flux [28, 30]. Binding studies have confirmed that they also have an affinity for the sigma receptor and that the polycyclic amine group is important for binding interactions [31-33]. They further have the ability to greatly improve lipophilicity of their conjugates, which is helpful in increasing blood-brain barrier permeability leading to increased concentration of CNS acting drugs in the brain, decreasing dosage and ultimately minimising peripheral side effects [34, 35]. Several studies have shown that both HCD containing compounds and the structurally related adamantane moiety may present with sigma receptor activity [31-33].
Based on the above observations a series of hexacyclododecylamines were designed and synthesised to evaluate the effect of including an amine-containing heterocyclic group compared to previous (HCD) structures containing only two lipophilic regions (Fig. 1 and Table 1). The inclusion of an amine-containing ring has been shown to improve σ1R activity in an array of structurally distinct compounds [36, 37]. This design would, therefore, serve as a preliminary study into the potential of including an amine-containing heterocyclic ring between the two lipophilic regions of these compounds. This may lead to better alignment of the compounds in the σ1R and/or additional binding interactions with the σ1R. In addition, σ1R antagonists have shown to regulate L-type VGCCs which may affect Ca2+ flux through these channels [12, 13]. The purpose of this study was thus to evaluate a series of HCD derivatives for modulating effect on the σ1R and intracellular calcium concentrations through VGCCs in an attempt to explore the neuromodulatory potential of this structural class of compounds.
2. EXPERIMENTAL SECTION
2.1. Chemistry: General Procedures
Unless otherwise specified, materials were obtained from Sigma Aldrich® and Merck® and used without further purification. All reactions were monitored by thin-layer chromatography on 0.20 mm thick aluminium silica gel sheets (Alugram® SIL G/UV254, Kieselgel 60, Macherey-Nagel, Düren, Germany). Visualisation was achieved using a Chromato-vue® Cabinet under UV light (254 nm and 366 nm) or with iodine vapours. Mobile phases were prepared on a volume-to-volume basis. Infra-Red (IR) spectra were recorded on a Perkin Elmer Spectrum 400 spectrometer, fitted with a diamond Attenuated Total Reflectance (ATR) attachment. Mass Spectra (MS) were recorded on an analytical VG 70-70E mass spectrometer using Electron Spray Ionisation (ESI) at 70 eV. 1H and 13C NMR spectra were obtained using a Varian Gemini 200 spectrometer at a frequency of 200 MHz and 50 MHz, respectively or a Bruker Advanced 600 Spectrometer at frequencies of 600 MHz and 150 MHz respectively. Tetramethylsilane (TMS) was used as a point of reference in all NMR experiments. All chemical shifts are reported in parts per million (ppm) relative to the signal from TMS (δ = 0), added to an appropriate deuterated solvent. All chemical shifts are reported in parts per million (ppm), relative to the internal standard. The following abbreviations are used to indicate the multiplicities of the respective signals: s – singlet; d - doublet; dd - doublet of doublets; t - triplet; m - multiplet; and AB-q - AB quartet. The multiplicity of the identified carbons was confirmed with DEPT-spectra. Microwave synthesis was performed using a CEM DiscoverTM microwave synthesis system. Compounds 6 [38], 8 [39], 9 [40], 13 [41], 15 [30], 21 [42] and 22 [40] were synthesised as previously described and all physical characteristics were similar to that reported in the literature.
2.2. Synthesis of Compounds
2.2.1. 5-[2-(4-Benzylpiperazin-1-yl)ethyl]-5-azahexacyclo[5.4. 1.02,6.03,10.04,8.09,12]dodecan4-ol (12)
Pentacyclo[5.4.0.02,6.03,10.05,9]undecane-8,11-dione-ethylene acetal (9, 200 mg, 0.917 mmol) and compound 8 (200 mg, 0.913 mmol) were dissolved in 5 ml EtOH and reacted under microwave conditions at a maximum temperature of 100°C, power setting of 150 W and pressure of 150 psi for 30 minutes. The reaction mixture was allowed to cool and directly used in the next step. The cooled solution of crude compound 10 was dissolved in 5 ml EtOH and NaBH4 (159 mg, 4.20 mmol, 1.4 equiv.) was added. The mixture was stirred at room temperature for 8 hours where after EtOH was evaporated under reduced pressure, water (10 ml) was added and the mixture was extracted with DCM (3 × 10 ml). The combined organic extracts were washed with brine (10 ml), dried (Na2SO4), and concentrated in vacuo. To this crude material (11), acetone (25 ml) and 4 M aq. HCl (15 ml) were added. After stirring at room temperature for 6 h, the mixture was diluted with H2O (200 ml), basified to pH 14 with 1 M aq. NaOH, and extracted with DCM (3 × 15 ml). The combined organic extracts were dried (Na2SO4) and concentrated in vacuo. The crude product was purified by recrystallization from EtOH to yield the desired compound (12) as colourless crystals (Yield = 183 mg, 53%) [38]. Physical data: 1H NMR (200 MHz, CDCl3) δH: 7.26 – 7.16 (m, 5H), 3.43 (s, 2H), 3.32 (t, 1H, J = 4.9 Hz), 2.96 – 2.30 (m, 20H), 1.79-1.41 (AB-q, 2H, J = 10.6 Hz). 13C NMR (50 MHz, CDCl3) δc: 137.9, 129.1, 128.2, 127.0, 70.8, 62.7, 58.6, 56.9, 52.7, 53.4, 51.5, 46.6, 45.7, 45.4, 43.3, 43.2, 42.1, 41.9, 41.7. IR (υmax): 3242.86, 2949.29, 2812.87, 1319.78, 1284.36 cm-1. HR-ESI [M+H]+: calcd. 378.2540, found. 378.2546.
2.2.2. N-[2-(4-benzylpiperidin-1-yl)ethyl]-5-oxahexacyclo[5.4. 1.02,6.03,10.04,8.09,12]dodecan-4-amine (18)
Methanesulfonyl chloride (261 mg, 2.28 mmol) dissolved in 10 ml of a 50:40:10 diethyl ether:DCM:triethylamine solvent system was added dropwise to a stirred solution of compound 15 (500 mg, 2.28 mmol, 1.00 equiv.) in 20 ml of the same solvent system under nitrogen gas on an external ice bath consisting of ice, acetone and sodium chloride which cooled the reaction mixture to approximately -8 °C. At the moment of addition, an exothermal reaction took place, gas was expelled and the reaction vessel’s temperature increased despite the external ice bath. The reaction was allowed to stir overnight at room temperature. The solvents were evaporated in vacuo and the mixture was dissolved in 10 ml of DCM and washed with 2 x 10 ml of brine. The combined organic fractions were dried over MgSO4, filtered and evaporated in vacuo to produce a dark yellow oil. The product ((16, 2-({5-oxahexacyclo [5.4.1.02,6.03,10.04,8.09,12]dodecan-4-yl}amino)ethyl methanesulfonate)) was confirmed to be pure enough for subsequent reactions (Yield = 193 mg, 20%). Physical data: 1H NMR (200 MHz, CDCl3): δH: 4.68 - 4.53 (t, 1H, J = 5.6 Hz), 3.57 (t, 2H, J = 5.8 Hz), 3.05 (t, 2H, J = 5.8 Hz), 2.90 – 2.32 (m, 9H), 1.95 – 1.46 (AB-q, 2H, J = 10.5). Next, 16 (193 mg, 0.65 mmol) was reacted with 4-benzylpiperidine (17, 114 mg, 0.65 mmol, 1.00 equiv.). The reactants were dissolved in 7 ml of acetonitrile and a spatula point (150 – 200 mg) of potassium carbonate was added. It was then reacted in a microwave reactor for 10 min at a maximum temperature of 120 °C, a pressure which fluctuated around 90 psi and power output between 50 W and 100 W. The reaction mixture was then dissolved in 10 ml of DCM and washed with 2 x 5 ml of water. Further purification by column chromatography was done starting with hexane as mobile phase and gradually incorporating ethyl acetate into the mobile phase at increments of 10%. The product eluded at 50% ethyl acetate:hexane, but still contained impurities. The eluded crude compound was dissolved in 10 ml of DCM and extracted with 3 x 10 ml 0.01 M HCl solution. The acidic phases were collected and made basic with a 0.1 M NaOH solution. The compound was then extracted with 3 x 30 ml of DCM. The organic fractions were collected, dried with MgSO4 overnight, filtered and dried in vacuo to yield the desired compound (18) as a viscous yellow-orange oil (Yield = 23 mg, 9%). Physical data: 1H NMR (200 MHz, CDCl3): δH: 7.38 – 7.19 (m, 5H), 4.62 (t, 1H, J = 5.6 Hz), 3.17 – 3.13 (d, 2H, J = 8.2 Hz), 2.79 – 2.40 (m, 21H), 1.90-1.48 (AB-q, 2H, J = 10.5). 13C NMR (50 MHz, CDCl3): δc: 136.8, 128.5, 127.4, 126.3, 82.6, 60.9, 55.0, 53.2, 51.9, 47.9, 47.6, 44.7, 44.5, 43.6, 43.4, 41.9, 41.7, 41.6, 41.3, 29.6. IR (υmax): 2961.45, 2861.88, 1739.92, 1341.79, 1008.16 cm-1. HR-ESI [M+H]2+: calcd. 378.2587, found. 378.2574.
2.2.3. 5-(3-{3-[(Piperidin-1-yl)methyl]phenoxy}propyl)-5-aza- hexacyclo[5.4.1.02,6.03,10.04,8.09,12]dodecan-4-ol (24)
Pentacyclo[5.4.0.02,6.03.10.05,9]undecane-8,11-dione (13, 1.4 g, 8.04 mmol) was reacted with 1.7 g (8.04 mmol) of N-[3-(3-piperidin-1-ylmethylphenoxy)propyl]amine (22) in 10 ml dry THF at -10 °C. The carbinolamine that formed after 10 min was filtered off and refluxed under Dean-Stark conditions for 1 hour in benzene. The benzene was evaporated under vacuum and the residue was dissolved in 6 ml dry methanol and 30 ml dry THF. To this, 0.3 g NaBH3CN was added as a reducing agent. The mixture was stirred overnight at room temperature. The methanol and THF were evaporated in vacuo and the residue was extracted with 4 x 25 ml DCM and the organic fraction was dried with MgSO4, filtered and evaporated in vacuo. The desired product (24) was obtained as a yellow oily substance after column chromatography using DCM: EtO Ac:PE, 1:1:1 (Rf = 0.42), as mobile phase (Yield: 0.43 g, 1.09 mmol, 13%). Physical data: 1H NMR (300 MHz, CDCl3) δH: 7.26 (t, 1H, J = 5.8 Hz), 6.95-6.88 (m, 1H), 6.80-6.74 (m, 2H), 4.10 (t, 2H, J = 5.1 Hz), 3.63 (t, 1H, J = 5.0 Hz), 3.52 (s, 2H) 3.13 (t, 2H, J = 5.2 Hz), 3.09-2.55 (m, 14H), 2.10-1.94 (m, 4H), 1.85-1.55 (AB-q, 2H, J = 11.3 Hz), 1.53-1.45 (m, 2H). 13C NMR (75 MHz, CDCl3) δc: 158.0, 140.2, 129.1, 122.0, 115.2, 113.0, 66.9, 64.8, 63.6, 54.8, 54.3, 54.3, 53.6, 46.3, 44.6, 43.9, 43.8, 43.1, 41.9, 41.6, 41.2 40.9, 38.9, 26.9, 26.0, 24.2 IR (υmax): 3342.8, 2969.1, 2865.4, 1731.0, 1337.7, 1274.1, 1098.0, 1557.9 cm-1. HR-ESI [M+H]: calcd. 407.2693, found. 407. 2697.
2.2.4. N-(3-{3-[(Piperidin-1-yl)methyl]phenoxy}propyl)-5-ox- ahexacyclo[5.4.1.02,6.03,10.04,8.09,12]dodecan-4-amine (25)
Pentacyclo[5.4.0.02,6.03.10.05,9]undecane-8,11-dione (13, 1.4 g, 8.04 mmol) was reacted with 1.7 g (8.04 mmol) of N-[3-(3-piperidin-1-ylmethylphenoxy)propyl]amine (22) in 10 ml dry THF at -10 °C. The carbinolamine that formed after 10 min was filtered off and refluxed under Dean-Stark conditions for 1 hour in benzene. The benzene was evaporated under vacuum and the residue was dissolved in 6 ml dry methanol and 30 ml dry THF. To this 0.30 g NaBH4 was added as reducing agent. The mixture was stirred overnight at room temperature. The methanol and THF were evaporated under vacuum and the residue was extracted with 4 x 25 ml DCM and the organic fraction was dried with magnesium sulphate, filtered and evaporated. The desired product (25) was obtained as an light yellow oily substance after column chromatography using DCM:EtOAc:PE, 1:1:1 (Rf = 0.43), as mobile phase. (Yield: 1.31 g, 35%). Physical data: 1H NMR (300 MHz, CDCI3) δH: 7.20 (t, 1H, J = 8.2 Hz), 6.93-6.85 (m, 2H), 6.78-6.74 (m, 1H), 4.7 (t, 1H, J = 5.4 Hz), 4.02 (t, 2H, J = 5.2 Hz), 3.43 (s, 2H), 3.17 (t, 2 H, J = 5.3 Hz), 3.10-3.01 (m, 4H), 2.96-2.65 (m, 10H), 2.15-2.09 (m, 4H), 2.05-1.87 (AB-q, 2H, J = 11.5), 1.60-1.53 (m, 2H); 13C NMR (75 MHz, CDCI3) δc: 159, 140.0, 128.1, 122.0, 115.4, 112.6, 110.0, 82.3, 65.2, 64.9, 64.0, 54.9, 54.8, 54.6, 54.0, 46.0, 44.5, 43.9, 43.5, 43.0, 41.5, 41.2 40.5 38.8, 26.8, 26.2, 24.2. IR (υmax): 3357.2, 2964.7, 2863.9, 1706.0, 1211.2, 1131.8 cm-1; HR-ESI [M+H]: calcd. 407.2693, found. 407.2696.
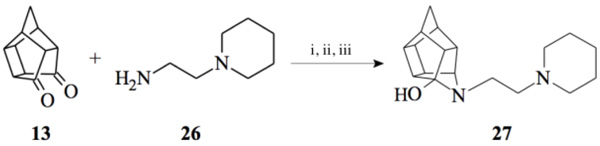
2.2.5. 5- [2- (Piperidin-1-yl) ethyl]-5- azahexacyclo [5.4.1.0 2,6. 03,10.04,8.09,12]dodecan-4-ol (27)
Pentacyclo[5.4.0.02,6.03.10.05,9]undecane-8,11-dione (13, 1 g, 8.04 mmol) was reacted with 1 ml of 1-(2-aminoethyl) piperidine (26, 5.74 mmol) in 10 ml dry THF at -10 °C. The carbinolamine that formed was filtered off and refluxed under Dean-Stark conditions for 1 hour in benzene. The benzene was evaporated in vacuo and the residue was dissolved in 6 ml dry methanol, 30 ml dry THF and 0.3 g NaBH3CN was added as reducing agent and the mixture was stirred overnight at room temperature. The methanol and THF were evaporated in vacuo and the residue was extracted with 4 x 25 ml DCM, dried with MgSO4, filtered and evaporated. The desired product (27) was obtained as a yellow oily substance after column chromatography using DCM:EtOAc:PE, 1:1:1 (Rf = 0.31), as mobile phase. (Yield: 0.70 g, 2.33 mmol, 41%). Physical data: 1H NMR (300 MHz, CDCl3) δH: 3.80 (t, 1H, J = 5.3 Hz), 2.80-2.10 (m, 16H), 1.90 (d, 1H, J = 10.40 Hz), 1.75-1.40 (m, 7H); 13C NMR (75 MHz, CDCI3) δc: 82.4, 71.9, 59.1, 58.1, 54.9, 54.8, 45.0, 44.8, 43.0, 42.6, 40.0, 35.2, 34.4, 30.5, 26.0, 24.6; IR (υmax): 3000, 2938.1, 2860.3, 2324.8, 2168.4 cm-1; HR-ESI [M+H]: calcd. 287.2118, found. 287.2121.
2.3. Sigma-1 Receptor Binding Affinity Studies
Binding affinities for the σ1R were determined as described previously [41, 42]. Briefly, rat liver lysates were incubated with 3 nM [3H](+)-pentazocine (Perkin Elmer USA), 100 ug/mL tissue lysate and compounds in 50 mM Tris pH 8.0. After 1 hour incubation, the reaction was terminated with the addition of 1 ml cold 50 mM Tris pH 8.0 buffer and filtered through Wathman B filter paper using a vacuum manifold. The filters were pre-soaked in 0.3% polyethylene imine for at least 30 min before the experiment was terminated. The filters were additionally washed twice with 1 ml cold buffer. The remaining radioactivity remaining on the filter paper was counted using scintillation counting. All data analysis, calculation and graphs were done using Prism 6.0® (GraphPhad, La Jolla, CA). Experiments were repeated three times on different tissue preparations with three determinations in each replicate. The final results were expressed as Ki and IC50 values.
2.4. Sigma-1 Receptor Docking Studies
The docking studies were performed using the recently published σ1R crystal structure (PDB: 5HK2) [43]. The Molecular Operating Environment (MOE) 2015 software suite was used for the docking studies with the following protocol. (1) The receptor protein structure was checked for missing atoms, bonds and contacts. (2) Hydrogens and partial charges were added using the protonate 3D application in MOE. Protein structure preparation was done by setting the pH of the protein at 7.4. (3) The ligands were constructed using the builder module and were energy minimized using CHARMm. (4) Ligands were docked within the σ1R active sites using MOEDock application. The active site was selected based on the position of the co-crystallised ligand, 4-IBP. The docking algorithm which was chosen for these experiments was based on induced fit docking to allow for flexible interactions of the test compounds with the receptor. (5) The top binding best pose of each compound was visually inspected and the interactions with binding pocket residues were analyzed. To determine the accuracy of this docking protocol, the co-crystallised ligand, 4-IBP (PDB ID: 5HK2), was redocked into the σ1R active site. The best ranked solution of 4-IBP exhibited an RMSD values of less than 0.56 Å from the position of the co-crystallised ligand. In general, RMSD values smaller than 2.0 Å, indicate that the docking protocol is capable of accurately predicting the binding orientation of the co-crystallised ligand [49, 50]. This protocol was thus deemed to be suitable for the docking of the test compounds.
2.5. Voltage Gated Calcium Channel Assay
The fluorescent ratiometric indicator, Mag-Fura-2/AM, and a Bio-Tek® fluorescent plate reader were used to evaluate the influence of the test compounds on calcium homeostasis via the VGCC utilizing murine synaptoneurosomes at 37 ºC. The preparation of synaptoneurosomes, solutions and experimental techniques was similar to those of published studies [44, 45]. All data analysis, calculation and graphs were conducted using Prism 6.0® (GraphPhad, La Jolla, CA). Data analysis was carried out using the Student Newman Keuls multiple range test and the level of significance was accepted at p < 0.05. Experiments were repeated three times on different tissue preparations with three determinations in each replicate
A 1990 µl suspension of synaptoneurosomes was allowed to reach room temperature, thereafter 10 µl of Fura-2 AM (1 mM in DMSO) was added to produce a final concentration of 5 µM. Synaptoneurosomes were then incubated at 37 ºC for 30 min after which the suspension was centrifuged on a desktop centrifuge at 7000 g for 5 min and the supernatant decanted to remove all extracellular Fura-2 AM. The resulting pellet was resuspended in 2 mM CaCl2 containing buffer to obtain a final protein concentration of 0.6 mg/ml.
For the screening test, 10 mM stock solutions of the compounds in DMSO were prepared, with the control containing 1% DMSO and no test compound. The individual stock solutions were added in 2 µl portions to a 96 well plate in triplicate followed by the addition of 200 µl of synaptoneurosomal-Fura-2 AM solution prepared above. This gave rise to a final concentration of 100 µM of the compounds. The 96 well plate was shaken and incubated for 30 min at 37 ºC and used immediately after incubation. The measurement was then performed at 37 ºC in a 96 well plate using dual wavelength excitation at 340 nm and 380 nm. The resting fluorescence was measured at 510 nm after which the changes in fluorescence intensity following the addition of 10 µl KCl (140 mM) depolarization solution using auto-injectors were recorded over a period of 5 min. The changes in fluorescence indicated the effect of the test compound on calcium flux. The data obtained from the fluorescent readings of each well were expressed in the form of a ratio (340 nm reading / 380 nm reading). This ratio indicated the net movement of Ca2+ ions across the membrane as it represents the ratio between unbound-Ca2+ and Ca2+-bound to Fura-2 AM. The average ratio over a 10 second interval before stimulation was subtracted from the average ratio 10 second after stimulation to give the net change in Ca2+ movement (Nc) across the membrane. This calculation was performed for each individual well and the averages of wells containing the same test compound were calculated (NcAve). These averages of three independent experiments were used to calculate the percentage inhibition of the test compounds relative to the control by the use of the following equation:
% inhibition = [NcAve (Control) – NcAve (Test compound)] / NcAve (Control) x 100%
3. RESULTS & DISCUSSION
3.1. Chemistry
To obtain the required novel oxa- and aza-hexacyclododecylamine compounds (12, 18, 24, 25 and 27), bimolecular nucleophilic substitution, nucleophilic addition and selective reduction methods were utilized. The synthesis of the aza-HCD compound 12 commenced from a series of microwave-assisted SN2 reactions to produce the intermediate 2-(4-benzylpiperazin-1-yl)ethanamine (8, Scheme 1), as previously described [38]. Compound 8 was conjugated to the pentacycloundecane (PCU) ketal (9) [39] through microwave irradiation at 100 °C, 150 W and 150 psi for 30 minutes to produce the PCU-imine (10). The PCU-imine was reduced using NaBH4 to produce the endo-amine (11) and the final aza-HCD (12) was obtained through hydrolysis and subsequent transannular cyclization using 3 M HCl/acetone.
To synthesize the oxa-HCD derivative (18), the obtained 2-aminoethanol-HCD structure (15, Scheme 2) [28] was reacted with methanesulfonyl chloride to provide a leaving group for reaction with the N-benzylpiperazine. Mesylate compounds generally show selectivity for amine groups over hydroxyl groups, but the NMR of compound 16 and the NMR, IR and MS data of 18 confirmed that the mesylate substitution of 16 took place on the hydroxyl moiety. This is probably due to steric hindrance from the hexacyclododecane structure at the amine. The benzyl piperidine (17) was then capable of displacing the methane-sulfonyl group and the final compound (18) was obtained using microwave-irradiation conditions (Scheme 2).
Compounds 21 and 22 were synthesised according to the methods proposed by Buschauer et al., (1985) [40]. Intermediate 22 served as the functional moiety that was conjugated to 13 in order to obtain imine-PCU compound 23. Further reduction using NaCNBH3 and NaBH4 produced the desired aza-HCD (24) and oxa-HCD (25) respectively (Scheme 3). Compound 27 was synthesised by conjugating 13 and 26 followed by Dean-Stark dehydration and subsequent reductive amination using NaCNBH3 (Scheme 4).
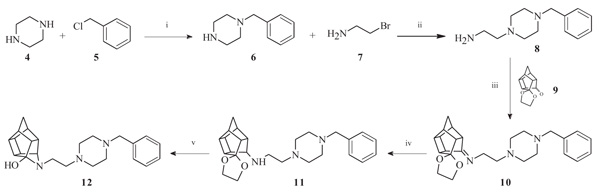
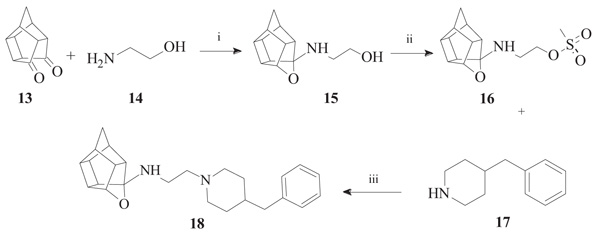
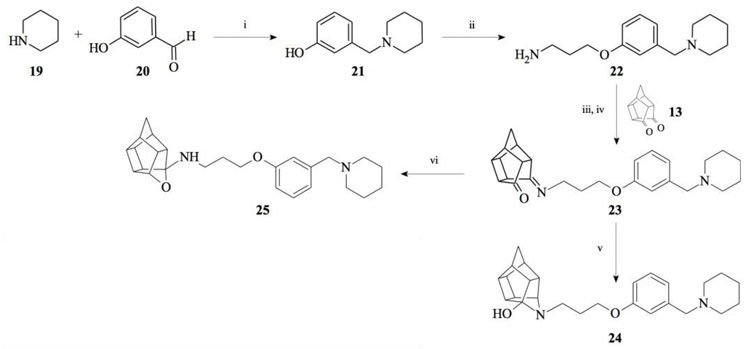
3.1.1. Sigma-1 Receptor Binding Affinity
σ1R binding affinity of the test compounds (Table 1) was evaluated according to the method by Matsumoto et al. In the experiments, rat liver membranes were used as the source of σ1Rs. [3H](+)-pentazocine was used as a radioligand and all stock solutions and procedures including data processing were adhered to as previously described [41]. The final results were expressed as Ki and IC50 values and further compared to known σ1R ligands. The novel HCD compounds (12, 18, 24, 25 and 27) showed σ1R affinities in the same range as that described for similar HCD structures (NGP1-01 and 1-3, Table 1) with 5-[2-(4-benzylpiperazin-1-yl)ethyl]-5- azahexacyclo [5.4.1.02,6 .03,10 .04,8.09,12] dodecan-4-ol (12) showing the best affinity (67 nM). Amongst the tested compounds, the aza structures generally are more favourable for σ1R binding than their oxa counterparts (cf. 12 and 18; 24 and 25). With the inclusion of the amine heterocycle between the cage and the aromatic ring (12 and 18) σ1R affinity was retained (cf. 12 with 2 and 3) or even improved (cf. 18 with NGP1-01 and 1) (Table 1). The addition of the HCD scaffold showed a definite increase in σ1R binding affinity from the respective intermediate counterparts without the HCD scaffold (cf. 8, 12 and 18; 22 and 24). Compounds 24 and 25 that consisted of the HCD connected to a phenyl ether with a terminal piperidine moiety, when compared to compounds 12 and 18, showed a reduced affinity and suggested that the HCD linked through an amine-containing heterocycle connected to a benzyl moiety is preferable for σ1R affinity and supports the pharmacophore model suggested by Glennon et al. [42] The aza compound 24 (Ki = 0.215 µM) compared to the oxa compound 25 (Ki = 11.855 µM) showed a significant increase in σ1R binding affinity which strongly suggests that the aza compound is preferable for the σ1R affinity. This preference for the aza-structure is further evident in the binding affinities observed for the aza-HCDs 2, 3, 12, 24 and 27. This is also in agreement with the previously published σ1R affinities for HCD derivatives [33, 39]. Intermediate compound 21 has good affinity (Ki = 0.349 µM) as it possesses the necessary features of the pharmacophore model including the basic amine and the adjacent lipophilic region which support this result. It is also important to note that intermediate compound 22 (Ki = 0.251 µM), despite the lack of a second lipophilic binding area, still showed significant σ1R binding. Finally, the comparison of compound 12 (0.067 µM) with compound 27 (Ki = 0.469 µM) indicates that the addition of N-benzyl group is favoured for σ1R affinity. When the activities of 12 and 27 are compared to 2 and 3, it is clear that the inclusion of the amine heterocycle leads to σ1R binding affinity in the same range. This suggests that the additional amine heterocycles retained the optimal alignment of compound 12 and 27 for σ1R binding affinity as suggested by the pharmacophore model proposed by Glennon et al. [42].
3.1.2. Sigma-1 Receptor Docking Studies
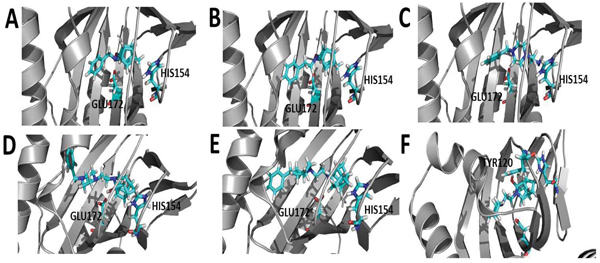
To gain insight into the mode of interaction between the compounds, σ1R, docking studies were performed using the Molecular Operating Environment (MOE) 2016 software package and the recently published crystal structure of the σ1R (PDB:5HK2) [43]. The docking results indicate that the HCU amine moiety seemed to play an important role in orienting the compounds for favorable hydrogen bonding interactions with several amino acids, specifically HIS154 and GLU172. For instance, compound 2, 3, 12 and 24 were found to form lipophilic interactions between the HCD carbons and HIS154, and the HC hydroxyl moiety with GLU172 (Fig. 2) through a hydrogen bond. Compound 18, an oxa-HCD showed an interaction with HIS154 but lacked the interaction with GLU172 as observed for the aza-HCDs 2, 3 and 12. This may explain why better σ1R affinity was observed for the aza-HCDs. Compound 8 without the HCD moiety did not show interactions with these two amino acids, which may account for the loss of affinity as compared to compounds 12 and 18 for instance. Compound 27, that lacks the benzyl group observed in 12 and 18, adopted an orientation where a hydrogen bond is observed between the hydroxyl moieties of the aza-HCD and TYR120. The Ki value of 27 was significantly higher compared to 12 and 18. This indicates that the benzyl moiety of 12 and 18 is important to enable optimal alignment of both the amine heterocycle and aza-HCD of these molecules within the σ1R binding pocket, enabling important interactions with HIS154 and GLU172.
3.2. Voltage-gated Calcium Channel Activity
All HCD amine compounds (Table 1) were screened at 100 µM for their potential inhibitory activity on the VGCC as previously described [44] using the fluorescent ratiometric indicator Fura-2 AM. Fresh synaptoneurosomes were prepared from rat brain homogenate [45] and incubated with Fura-2 AM. Thereafter the test compounds were incubated for 30 minutes and 140 mM KCl solution was added to depolarize the cell membranes to stimulate calcium influx. Calcium influx was then monitored based on the fluorescence intensity relative to that of a blank control (without inhibiting compound) over a 5 minute period. Two positive controls were included in the VGCC assay; nimodipine, a commercially available dihydropyridine calcium channel blocker and NGP1-01, the prototype HCD compound. Amantadine, a polycyclic NMDA receptor channel inhibitor, was also included as a negative control. Dose-response curves were plotted for selected compounds which showed a high degree of VGCC inhibition at 100 µM. The novel aza derivatives 24 and 27 exhibited comparable activities (IC50 < 10 µM) in the KCl initiated calcium flux assays to NGP1-01 (IC50 = 86 µM) and related HCD’s 1-3 (IC50 = 8 – 51 µM). The aza-HCD compound (12) showed moderate calcium flux inhibition (IC50 < 100 µM) and the two oxa-HCD derivatives 18 and 25 exhibited the weakest calcium flux inhibition (26% and 25%) at 100 µM (Table 1). From the VGCC results compounds 12, 24 and 27 can be considered as lead compounds to develop more potent calcium channel inhibitors.
| Number | Structure | σ1R Affinity | % Ca2+ Flux Inhibition KCl [100 µM] |
|
|---|---|---|---|---|
| Ki (µM) | IC50 (µM) | |||
| Amantadine |  |
7.44 [46] | 15.24 | 3% |
| NGP1-01 |  |
0.869 [33] | 1.78 | 52%** IC50 = 86 uM |
| 1 |  |
0.752 [33] | 1.54 | 89%*** IC50 = 51 uM |
| 2 | 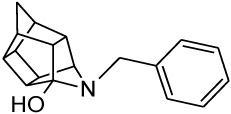 |
0.103 [33] | 0.211 | > 95%*** IC50 = 35 uM |
| 3 | 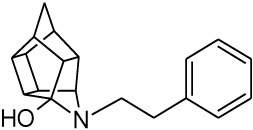 |
0.020 [33] | 0.041 | > 95%*** IC50 = 8 uM |
| 8 | 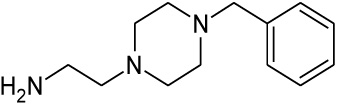 |
1.132 | 2.34 | nd |
| 12 | 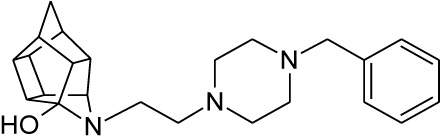 |
0.067 | 0.139 | 51%** IC50 = < 100 µM |
| 18 | 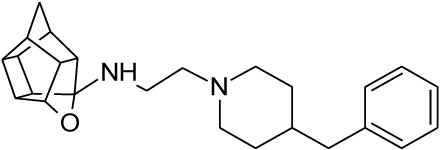 |
0.132 | 0.273 | 26% |
| 21 | 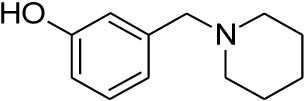 |
0.349 | 0.723 | nd |
| 22 | 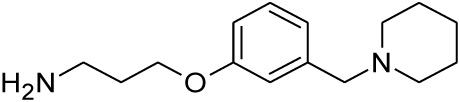 |
0.251 | 0.518 | nd |
| 24 | 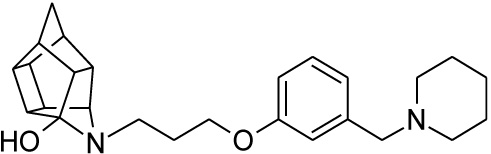 |
0.215 | 0.445 | 97%*** IC50 = 9.28 μM |
| 25 | 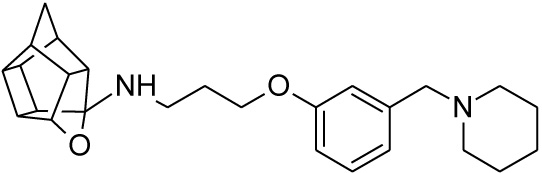 |
11.855 | 24.5 | 25% |
| 27 | 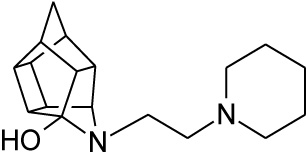 |
0.469 | 0.971 | 79%*** IC50 = 9.80 μM |
| Nimodipine | - | nd | nd | > 95%*** IC50 = 0.008 μM |
CONCLUSION
The synthesised HCD derivatives had inhibitory effects on calcium flux and thus decreased intracellular calcium concentration. In many cases, this effect might be linked to the compounds’ interaction with sigma receptors present in the synaptoneurosomal preparation and implicates σ1R antagonism. This is corroborated by the correlation observed between the VGCC inhibitory activity and the σ1R affinity of NGP1-01, 1, 2, 3 and between 12 and 18, and 24 and 25 – as the σ1R affinity for these compounds increased, the VGCC inhibition activity also increased. The decrease in intracellular calcium detected in the VGCC assays could thus in part be linked to the interaction of these compounds with σ1R in this assay. The compounds where this correlation is not observed will also be useful in identifying structural features to manipulate the specific or dual activities of future structures in this series. The observations and conclusions obtained from this study do not exclude interaction with other receptors such as the NMDA and AMPA/KA receptors and further investigation to characterise the calcium modulating effects of these compounds will follow.
Test compounds 12, 24 and 27 show potential to act as dual σ1R antagonists and VGCC blockers. Compound 12 exhibited VGCC inhibition in the same range as NGP1-01 with 24 and 27 showing activity comparable to the most potent aza-HCD compound 3 (Table 1). All three of these compounds showed nanomolar σ1R binding affinity (Ki = 0.067 – 0.469 µM). The dual and possible multifunctional activity of these compounds justifies further biological evaluation to establish their potential as multifunctional neurotherapeutic agents. A further important consideration when comparing the HCDs affinity to other σ1R ligands is their ability to cross the blood-brain barrier [34, 35] and other relevant neuroprotective /neurological activities, providing a multitarget-directed drug approach. This study has also shown that the inclusion of an amine heterocycle into these structures could be a viable option in the design of new σ1R ligands with VGCC activity. The reported compounds could prove to be effective lead structures in the development of therapies for the prevention and treatment of neurodegenerative and neurological disorders.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
No animals/humans were used for studies that are the basis of this research.
CONSENT FOR PUBLICATION
Not applicable.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
The authors would like to thank the National Research Foundation and Medical Research Council of South Africa for research funding. The project described was supported by the National Institute Of General Medical Sciences (NIH: NIG MS), U54GM104942. The content is solely the responsibility of the authors and does not represent the official views of the funding bodies.

