All published articles of this journal are available on ScienceDirect.

Design and Synthesis of WM5 Analogues as HIV-1 TAR RNA Binders
Authors Info & Affiliations
Abstract
Background:
The 6-aminoquinolone WM5, previously identified by us, is among the most selective small molecules known as TAR RNA binders to show anti-HIV activity.
Methods:
Starting from WM5, a series of analogues modified at N-1, C-6 or C-7 position was prepared by inserting guanidine or amidine groups as well as other protonable moieties intended to electrostatically bind the phosphate backbone of TAR. All the compounds were tested for their ability to inhibit HIV-1 replication in MT-4 cells and in parallel for their cytotoxicity. The active compounds were also evaluated for their ability to interfere with the formation of the Tat-TAR complex using a Fluorescence Quenching Assay (FQA).
Results:
Some of the synthesized compounds showed an anti-HIV-1 activity in the sub-micromolar range with the naphthyridone derivatives being the most potent. Three of the synthesized derivatives were able to interact with the Tat-TAR complex formation presenting Ki values improved as compared to the values obtained with WM5.
Conclusion:
The addition of a pyridine-based protonable side chain at the N-1 position of the quinolone/naphthyridone core imparted to the compounds the ability to interfere with Tat-TAR complex formation and HIV-1 replication.
1. INTRODUCTION
In the past decades, considerable work has been done in exploiting small non-coding hairpin RNA fragments as drug targets. However, compared with the compounds targeting proteins, the identification of molecules that selectively bind to structured RNA motifs remains an important challenge [1-3]. One of the most studied RNA structures is the Transactivation response element (TAR) of HIV-1 genome [4-9], a short stem-bulge-loop structure located in the long terminal repeat region at the 5’ end of all nascent HIV-1 transcripts. Its specific interaction with the HIV-1 regulatory protein Tat is essential for viral gene expression, replication and pathogenesis [10, 11].
TAR RNA is extremely conserved among viral isolates making it a very attractive target. During the last two decades, many small molecules, aminoglycosides-conjugates, Tat-based peptidomimetics, and peptides have been reported as TAR binders [12-19].
Among the small molecules known as TAR RNA binder, the 6-aminoquinolone WM5 Fig. (1), which was developed in our lab, inhibits the Tat-mediated transactivation by interfering selectively with the bulge region of TAR RNA [20].
Numerous derivatives have been designed and synthesized starting from WM5 with the aim to gain Structure-Activity Relationship (SAR) insights, reduce the cytotoxicity and further investigate the molecular mechanism of action [21]. As a consequence, many compounds endowed with good HIV-1 and HIV-2 inhibitory activity in both chronically infected and acutely infected cells have already been identified. However, a direct correlation between the anti-HIV activity and their ability to interfere with the Tat-TAR RNA complex was not always observed [22]. On the other hand, when the quinolone nucleus lacking both C-7 arylpiperazine and C-3 carboxylic functions was properly functionalized with protonable groups at the C-2 position, a series of 2-phenylquinolones was obtained showing an improved ability to displace Tat-TAR complex but at the expense of antiviral activity [23, 24].
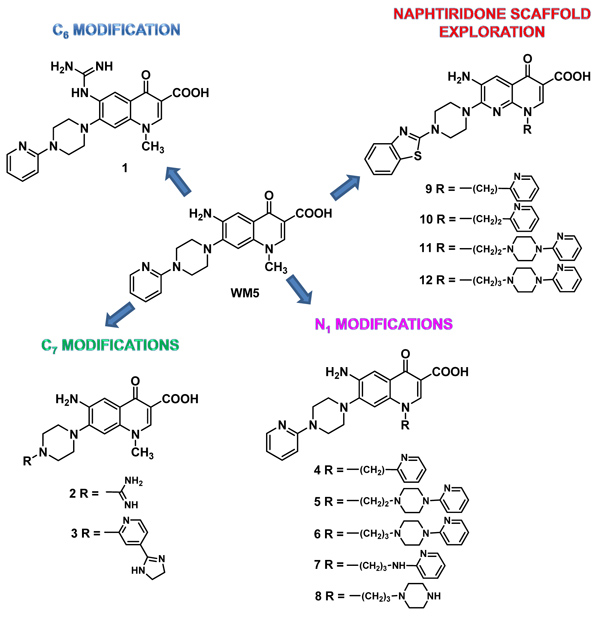
Here, we came back to WM5 preparing a series of analogues designed to enhance its ability to recognize the viral nucleic acid TAR RNA. Considering that TAR plays its essential role in the Tat-mediated transcription by specifically binding Tat and that in the Tat-TAR complex the arginine rich basic portion of Tat is responsible for the TAR binding, we decided to modify WM5 by inserting guanidine or amidine groups as well as other protonable moieties intended to bind electrostatically the phosphate backbone of TAR. In the present paper, the synthesis of a series of WM5 analogues modified at N-1, C-6 or C-7 position Fig. (1), and their anti-HIV-1 activity along with their cytotoxicity in MT-4 cells, are reported. The active compounds were also evaluated in a Fluorescence Quenching Assay (FQA) to determine their ability to interfere with the formation of the complex between TAR RNA and a truncated Tat peptide.
1.1. Design of WM5 Analogues
Starting from WM5, the C-6 amino group was replaced by a more basic guanidine in compound 1, while at C-7 position an amidine moiety replaced the 2-pyridine ring in compound 2, and an imidazoline ring was present at the C-4 position of the pyridine in compound 3 Fig. (1). Pyridine and (pyridinyl)piperazine moieties were inserted at the N-1 position through methyl, ethyl, or propyl chains, as in compounds 4, 5 [25] and 6, respectively Fig. (1). Moreover, by maintaining the propyl chain, the piperazine and the pyridine rings were alternatively deleted as in compounds 7 and 8, respectively Fig. (1). We have previously reported that the 1,8-naphthyridone nucleus, when decorated with a 1,3-benzothiazolpiperazine moiety at the C-7 position, grants potent and selective anti-HIV activity [26, 27]. Thus, the same flexible protonable side chains were also inserted at N-1 position of the 1,8-naphthyridone scaffold, obtaining compounds 9 [25], 10 [25], 11, and 12 Fig. (1).
2. MATERIALS AND METHODS
2.1. Chemistry
Starting materials, reagents, and solvents that were commercially available were used as supplied. The reactions were monitored by TLC on silica gel 60F254 (Merck) and the compounds were visualized by UV and/or iodine. Flash chromatography columns were performed on Merck silica gel 60 (mesh 230-400). After extraction, organic solutions were dried using anhydrous Na2SO4, filtered, and evaporated to dryness at reduced pressure using a Büchi rotary evaporator. Yields are of pure products and were not optimized. Melting points were determined in capillary tubes (Büchi Electrothermal Mod. 9100) and are uncorrected. Elemental analyses were performed on a Fisons elemental analyzer, Model EA1108CHN, and the data for C, H and N are within ± 0.4% of the theoretical values. 1H NMR and 13C NMR spectra were recorded at 200 MHz (Bruker Avance DPX-200) and 400 MHz (Bruker Avance DRX-400) using residual solvents such as dimethylsulfoxide (δ = 2.48) or chloroform (δ = 7.26) as an internal standard. Chemical shifts (δ) are reported in parts per million (ppm), and peak multiplicity are reported as s (singlet), d (doublet), t (triplet), q (quartet), p (pentet), hept (heptet), m (multiplet), or brs (broad singlet). Target compounds 5 [25], 9 [26], 10 [27] have been synthesized as previously reported.
2.1.1. 6-(E)-(2,3-Di-Tert-Butoxyguanidino)-1-Methyl-4-Oxo- 7-[4-(Pyridin2-yl)Piperazin-1-yl]]-1,4-Dihydroquinoline-3-Carboxylic Acid (13)
To a solution of WM5 [20] (0.13 g, 3.29 mmol) in dry DMF (10 mL), Et3N (0.07 g, 7.24 mmol, 0.1 mL), N,N′-di-(tert-butoxycarbonyl)thiourea (0.10 g, 3.45 mmol), and HgCl2 (cat. amount) were added. The mixture was reacted at room temperature for 15 min. The solvent was subsequently concentrated under vacuum and the residue was treated with EtOH yielding a precipitate that was eliminated by filtration. The filtrate was evaporated to dryness obtaining a solid that was washed with hot EtOH (3 times), filtered off, and dried to give compound 13 (0.10 g, 10%): mp > 300 °C; 1H NMR (400 MHz, CDCl3) δ 1.50 (s, 18H, CH3), 3.05-3.25 (m, 4H, piperazine CH2), 3.70-3.90 (m, 7H, CH3 and piperazine CH2), 6.60-6.75 (m, 2H, pyridine CH), 7.05 (s, 1H, H-8), 7.40 (bs, 1H, NH), 7.50 (t, J = 6.0 Hz, 1H, pyridine CH), 8.10 (d, J = 6.0 Hz, 1H, pyridine CH), 8.70 (s, 1H, H-5), 9.40 (s, 1H, H-2), 10.80 (bs, 1H, NH), 12.40 (s, 1H, COOH).
2.1.2. 6-Guanidino-1-Methyl-4-Oxo-7-(4-(Pyridin2-yl) Piperazin -1-yl])-1,4-Dihydroquinoline-3-Carboxylic Acid (1)
HCl was bubbled through a solution of compound 13 (0.10 g, 1.74 mmol) in a mixture of absolute EtOH (10 mL) and CHCl3 (10 mL), at room temperature. After 15 h, the obtained precipitate was collected by filtration, washed with a mixture of EtOH/CHCl3 (1:1), and dried, to give 1 (0.018 g, 26%): mp > 300 °C; 1H NMR (400 MHz, DMSO-d6) δ 3.10-3.25 and 4.10-4.25 (m, each 4H, piperazine CH2), 3.90 (s, 3H, CH3), 7.00 (t, J = 6.4 Hz, 1H, pyridine CH), 7.30 (s, 1H, H-8), 7.50 (d, J = 9.5 Hz, 1H, pyridine CH), 8.05-8.10 (m, 2H, pyridine CH), 8.25 (bs, 1H, NH), 8.75 (s, 1H, H-5), 8.90 (s, 1H, H-2), 9.50 (bs, 1H, COOH). Anal. calcd. for C21H22N6O3: C 59.85, H 5.50, N 23.26, found: C 60.00, H 5.75, N 23.00.
2.1.3. Ethyl 7-(4-Carbamidoylpiperazin-1-Yl)-1-Methyl-6-Nitro-4-Oxo-1,4-Dihydroquinoline-3-Carboxylate (15)
To a solution of synthon 14 [28] (0.70 g, 1.92 mmol) in dry DMSO (50 mL), methyl imidothiocarbamate sulfate (0.26 g, 0.959 mmol) was added. The reaction mixture was stirred at 120 °C for 6 h. After cooling, the precipitate obtained was filtered and dried, to give 15 (0.77 g, 99%): mp 179-181 °C; 1H NMR (400 MHz, DMSO-d6) δ 1.25 (t, J = 7.0 Hz, 3H, CH2CH3), 3.20-3.30 and 3.60-3.70 (m, each 4H, piperazine CH2), 3.90 (s, 3H, CH3), 4.25 (q, J = 7.0 Hz, 2H, CH2CH3), 7.15 (s, 1H, H-8), 8.10 (bs, 3H, NH and NH2), 8.60 (s, 1H, H-5), 8.70 (s, 1H, H-2).
2.1.4. 6-Amino-7-(4-Carbamidoylpiperazin-1-Yl)-1-Methyl-4-Oxo-1,4-Dihydroquinoline-3-Carboxylic Acid (2)
To a solution of 15 (0.15 g, 3.69 mmol) in H2O (200 mL) and NH4OH conc (5 mL), a solution of FeSO4 (0.71 g, 2.58 mmol) in the minimum amount of H2O was added and the mixture was stirred at room temperature. After 1 h, the reaction mixture was filtered over Celite® and the filtrate was evaporated to dryness to give a residue that was crystallized from H2O yielding ethyl 6-amino-7-(4-carbamimidoylpiperazin-1-yl)-1-methyl-4-oxo-1,4-dihydroquinoline-3-carboxylate (0.08 g, 58%), which was used in the next step without further purification. Thus, ethyl 6-amino-7-(4-carbamimidoylpiperazin-1-yl)-1-methyl-4-oxo-1,4-dihydroquinoline-3-carboxylate (0.08 g, 0.21 mmol) was dissolved in EtOH (3 mL) and 6N HCl (3 mL) was added. The reaction mixture was stirred at reflux for 7 h. After cooling, the precipitate obtained was filtrated, washed with EtOH, and purified by crystallization from MeOH/DMF, to give 2 (0.013 g, 17%): mp > 300 °C; 1H NMR (400 MHz, DMSO-d6) δ 2.90-3.20 and 3.60-3.90 (m, each 4H, piperazine CH2), 3.90 (s, 3H, CH3), 7.20 (s, 1H, H-8), 7.50 (s, 1H, H-5), 7.90 (bs, 3H, NH e NH2), 8.80 (s, 1H, H-2). Anal. calcd. for C16H20N6O3: C 55.80, H 5.85, N 24.40, found: C 55.63, H 5.98, N 24.35.
2.1.5. 1-[4-(4,5-Dihydro-1h-Imidazol-2-Yl)Pyridin-2-Yl]Piperazine (18)
A mixture of 16 [29] (0.50 g, 26.1 mmol), ethylenediamine (0.17 g, 2.87 mmol), and p-toluenesulfonic acid (0.54 g, 2.87 mmol) was reacted under reflux for 3 h. After cooling, the reaction mixture was diluted with H2O and then extracted with CHCl3. The organic layers were dried and evaporated to dryness to give 18 (0.40 g, 66%) as an oil; 1H NMR (400 MHz, DMSO-d6) δ 2.70-2.80 and 3.20-3.30 (m, each 4H, piperazine CH2), 3.65 (bs, 4H, imidazoline CH2), 3.80 (bs, 1H, NH), 7.00 (d, J = 5.0 Hz, 1H, pyridine CH), 7.20 (s, 1H, pyridine CH), 8.20 (s, 1H, pyridine CH).
2.1.6. General Procedure for the C-7 Nucleophilic Substitution Reaction (Method A)
A mixture of the selected synthon (1.0 equiv), the appropriate base (3.0 equiv), and K2CO3 (3.0 equiv) in dry DMF was stirred at 40-80°C until no starting material was detected by TLC (2-72 h). After cooling, the reaction mixture was poured into ice/water yielding a precipitate which was washed with water and then with Et2O, and further purified as reported in the description of the compounds.
2.1.7. Ethyl 7-(4-(4-(4,5-Dihydro-1h-Imidazol-2-Yl)Pyridin2-yl)Piperazin-1-yl])-1-Methyl-6-Nitro-4-Oxo-1,4-Dihydroquinoline-3-Carboxylate (19)
The title compound was obtained starting from 17 [20] and 18 using Method A (80 °C, 6 h) after crystallization from DMF in 88% yield: mp 255-256 °C; 1H NMR (400 MHz, DMSO-d6) δ 1.25 (t, J = 7.0 Hz, 3H, CH2CH3), 3.30-3.40 and 3.70-3.75 (m, each 4H, piperazine CH2), 3.80 (bs, 4H, imidazoline CH2), 3.95 (s, 3H, CH3), 4.25 (q, J = 7.0 Hz, 2H, CH2CH3), 7.05 (d, J = 5.0 Hz, 1H, pyridine CH), 7.10-7.15 (m, 1H, pyridine CH), 7.20 (s, 1H, H-8), 8.20 (d, J = 5.0 Hz, 1H, pyridine CH), 8.55 (s, 1H, H-2), 8.70 (s, 1H, H-5).
2.1.8. General Procedure for Reduction of C-6 Nitro Group (Method B)
A stirred solution of the selected 6-nitroderivative in DMF was hydrogenated over a catalytic amount of Raney nickel under atmospheric pressure at room temperature until no starting material was detected by TLC (15 min-3 h). The mixture was then filtered over Celite, and the filtrate was evaporated to dryness to afford a residue that was treated with EtOH/Et2O yielding a solid that was filtered and dried.
2.1.9. Ethyl 6-Amino-7-(4-(4-(4,5-Dihydro-1h-Imidazol-2-Yl)Pyridin2-yl)Piperazin-1-yl])-1-Methyl-4-Oxo-1,4-Dihydroquinoline-3-Carboxylate (20)
The title compound was obtained starting from 19 using Method B (3 h) after purification by flash chromatography eluting with CH2Cl2/MeOH (9:1) in 58% yield: mp 300 °C (dec); 1H NMR (200 MHz, DMSO-d6) δ 1.25 (t, J = 7.0 Hz, 3H, CH2CH3), 3.05-3.10 and 3.50-3.55 (m, each 4H, piperazine CH2), 3.80 (bs, 4H, imidazoline CH2), 3.90 (s, 3H, CH3), 4.20 (q, J = 7.0 Hz, 2H, CH2CH3), 5.25 (bs, 2H, NH2), 7.00 (s, 1H, H-8), 7.15 (d, J = 5.0 Hz, 1H, pyridine CH), 7.45 (s, 1H, H-5), 7.60-7.65 (m, 1H, pyridine CH), 8.30 (d, J = 5.0 Hz, 1H, pyridine CH), 8.40 (s, 1H, H-2).
2.1.10. General Procedure for Basic Hydrolysis (Method C)
A suspension of selected ethyl ester (0.3 mequiv) in 4% NaOH (5 mL) was refluxed until no starting material could be detected by TLC (4-20 h). After cooling the reaction mixture was acidified to pH = 4 with 2N HCl, obtaining a precipitate which was filtered, washed with water, and purified as described below.
2.1.11. 6-Amino-7-{4-[4-(4,5-Dihydro-1h-Imidazol-2-Yl) Pyridin -2-Yl]Piperazin-1-Yl}-1-Methyl-4-Oxo-1,4-Dihydroquinoline-3-Carboxylic Acid (3)
The title compound was obtained starting from 20 using Method C (15 h) after crystallization from DMF in 32% yield: mp > 300 °C; 1H NMR (400 MHz, DMSO-d6) δ 2.80-2.90 and 3.00-3.25 (m, each 4H, piperazine CH2), 3.50 (bs, 4H, imidazoline CH2), 3.90 (s, 3H, CH3), 5.00 (bs, 1H, NH), 5.50 (bs, 2H, NH2), 6.90 (s, 1H, H-8), 7.10-7.25 (m, 2H, pyridine CH), 7.50 (s, 1H, H-5), 8.25 (d, J = 5.0 Hz, 1H, pyridine CH), 8.75 (s, 1H, H-2), 11.20 (COOH). Anal. calcd. for C23H25N7O3: C 61.73, H 5.63, N 21.91, found: C 61.98, H 5.82, N 21.73.
2.1.12. General Procedure for the Cycloaracylation Reaction, Step 1 (Method D)
A mixture of acrylate (1.0 equiv) and appropriate amine (1.2 equiv) in a mixture of Et2O/EtOH (3:1), was stirred at room temperature until no starting material was detected by TLC (30 min-2 h). The reaction mixture was then concentrated in vacuo, yielding a residue which was used in the consecutive step without further purification.
2.1.13. Ethyl 2-(2,4-Dichloro-5-Nitrobenzoyl)-3-(Pyridin-2-Ylmethylamino)Acrylate (22)
The title compound was obtained starting from 21 [30] and (pyridin-2-ylmethyl)amine using Method D (2 h) in 78% yield: mp 98-99 °C; 1H-NMR (200 MHz, CDCl3) δ 1.00 (t, J = 7.1 Hz, 3H, CH2CH3), 4.00 (q, J = 7.1 Hz, 2H, CH2CH3), 4.75 (d, J = 5.9 Hz, 2H, CH2), 7.15-7.25 (m, 2H, pyridine CH), 7.50 (s, 1H, H-3’), 7.70-7.80 (m, 2H, pyridine CH and H-6’), 8.30 (d, J = 14.0 Hz, 1H, vinyl CH), 8.65-8.75 (m, 1H, pyridine CH), 11.40-11.50 (m, 1H, NH).
2.1.14. Ethyl 3-[(3-Bromopropyl)Amino]-2-(2,4-Dichloro-5-Nitrobenzoyl)Acrylate (23)
The title compound was obtained starting from 21 [30] and (3-bromopropyl)amine using Method D (30 min) as keto-enolic mixture in 66% yield: mp 66-67 °C; 1H-NMR (200, MHz, DMSO-d6) δ 0.75 and 0.90 (t, J = 7.0 Hz, 0.75 and 2.25H, CH2CH3), 2.10-2.25 (m, 2H, CH2), 3.50-3.75 (m, 4H, CH2), 3.90 (q, J = 7.0 Hz, 2H, CH2CH3), 4.20 (bs, 1H, NH), 8.10 (s, 2H, H-3’ and H-6’), 8.20-8.25 (m, 1H, vinyl CH), 9.75 (bs, 0.75H, OH), 10.90 (bs, 0.25H, NH).
2.1.15. Ethyl 2-(2,4-Dichloro-5-Nitrobenzoyl) -3-{[3-(Pyridin -2 -Ylamino)Propyl]Amino}Prop-2-Enoate (24)
The title compound was obtained as an oil, starting from 21 [30] and N-(pyridine-2-yl)propane-1,3-diamine [31] using Method D (30 min) in 29% yield; 1H-NMR (200 MHz, CDCl3) δ 1.05 (t, J = 7.1 Hz, 3H, CH2CH3), 1.90-2.00 (m, 2H, CH2CH2CH2), 3.50-3.60 (m, 4H, CH2CH2CH2), 3.95-4.05 (q, J = 7.1 Hz, 2H, CH2CH3), 4.50 (bs, 1H, NH) 6.35-6.45 (m, 1H, pyridine CH), 6.60-6.70 (m, 1H, pyridine CH), 7.30-7.40 (m, 1H, pyridine CH), 7.55 (s, 1H, H-3’), 8.75 (s, 1H, H-6’), 8.20-8.30 (m, 2 H, vinyl CH and pyridine CH), 11.25 (bs, 1 H, NH).
2.1.16. General Procedure For The Cycloaracylation Reaction, Step 2 (Method E)
A mixture of acrylate intermediate (1.0 equiv) and K2CO3 (3.0 equiv) in dry DMF was heated at 80°C for 1 h. After cooling, the reaction mixture was poured into ice/water, giving a precipitate that was filtered, washed with water and then with Et2O.
2.1.17. Ethyl 7-Chloro -6-Nitro-4 -Oxo-1- (Pyridin -2-Ylmethyl) -1,4- Dihydroquinoline- 3-Carboxylate (25)
The title compound was obtained starting from 22 using Method E in 86% yield: mp 228-230 °C; 1H NMR (400 MHz, CDCl3) δ 1.30 (t, J = 7.1 Hz, 3H, CH2CH3), 4.45 (q, J = 7.1 Hz, 2H, CH2CH3), 5.40 (s, 2H, CH2), 7.20-7.30 (m, 2H, pyridine CH), 7.55 (s, 1H, H-8), 7.65-7.75 (m, 1H, pyridine CH), 8.50-8.60 (m, 2H, pyridine CH and H-2), 8.95 (s, 1H, H-5).
2.1.18. Ethyl 1-(3-Bromopropyl)-7-Chloro-6-Nitro-4-Oxo-1,4-Dihydroquinoline-3-Carboxylate (26)
The title compound was obtained starting from 23 using Method E in 88% yield: mp 180-181°C; 1H NMR (400 MHz, DMSO-d6) δ 1.30 (t, J = 7.0 Hz, 3H, CH2CH3), 2.15-2.30 (m, 2H, CH2CH2CH2), 3.70 (t, J = 7,5 Hz, 2H, NCH2) 4.25 (q, J = 7.0 Hz, 2H, CH2CH3), 4.50-4.60 (m, 2H, CH2Br), 8.25 (s, 1H, H-8), 8.75 (s, 1H, H-5), 8.80 (s, 1H, H-2).
2.1.19. Ethyl 7- Chloro- 6-Nitro-4-Oxo-1- [3-(Pyridin-2-Ylamino) Propyl]-1,4- Dihydroquinoline-3- Carboxylate (27)
The title compound was obtained starting from 24 using Method E in 77% yield: mp 148-149 °C; 1H NMR (400 MHz, CDCl3) δ 1.40 (t, J = 7.1 Hz, 3H, CH2CH3), 2.20-2.30 (m, 2H, CH2CH2CH2) 3.50-3.60 (m, 2H, NCH2), 4.30-4.45 (m, 4H, CH2NH and CH2CH3), 4.60-4.70 (m, 1H, NH), 6.40-6.50 and 6.55-6.65 and 7.40-7.50 (m, each 1H, pyridine CH), 7.75 (s, 1H, H-8), 8.00-8.10 (m, 1H, pyridine CH), 8.60 (s, 1H, H-2), 9.00 (s, 1H, H-5).
2.1.20. Ethyl 6- Nitro -4-Oxo-1- (Pyridin-2-Ylmethyl) -7-(4-(Pyridin2-yl) Piperazin-1-yl]) -1,4- Dihydroquinoline -3- Carboxylate (28)
The title compound was obtained starting from 25 and 1-(2-pyridinyl)piperazine using Method A (80°C, 2 h) in 88% yield: mp 181-183 °C; 1H NMR (400 MHz, CDCl3) δ 1.35 (t, J = 7.0 Hz, 3H, CH2CH3), 3.05-3.15 and 3.55-3.65 (m, each 4H, piperazine CH2), 4.45 (q, J = 7.0 Hz, 2H, CH2CH3), 5.35 (s, 2H, CH2), 6.60-6.70 (m, 2H, pyridine CH), 6.90 (s, 1H, H-8), 7.15-7.30 (m, 2H, pyridine CH), 7.40-7.50, 7.60-7.70, and 8.15-8.25 (m, each 1H, pyridine CH), 8.60-8.65 (m, 2H, H-2 and pyridine CH), 8.80 (s, 1H, H-5).
2.1.21. Ethyl 6-Nitro-4-Oxo-7-(4-(Pyridin2-yl)Piperazin-1-yl])-1-[3-(4-(Pyridin2-yl)Piperazin-1-yl])Propyl]-1,4-Dihydroquinoline-3-Carboxylate (29) And Ethyl 1-(3-Bromopropyl)-6-Nitro-4-Oxo-7-(4-(Pyridin2-yl)Piperazin-1-yl])-1,4-Dihydroquinoline-3-Carboxylate (30)
The title compounds were obtained starting from 26 and 1-(2-pyridinyl)piperazine using Method A (40°C, 72 h) after purification by flash chromatography eluting with CHCl3/ MeOH (from 100:0 to 80:20) in 53% yield (compound 29) and 23% yield (compound 30); compound 29: mp 89-91 °C; 1H NMR (400 MHz, CDCl3) δ 1.40 (t, J = 7.0 Hz, 3H, CH2CH3), 2.05-2.10 and 2.40-2.45 (m, each 2H, CH2), 2.50-2.60, 3.25-3.35, 3.50-3.60 and 3.75-3.85 (m, each 4H, piperazine CH2), 4.25-4.45 (m, 4H, CH2CH3 and CH2), 6.60-6.75 (m, 4H, pyridine CH), 6.90 (s, 1H, H-8), 7.45-7.55 and 8.15-8.25 (m, each 2H, pyridine CH), 8.60 (s, 1H, H-5), 8.80 (s, 1H, H-2); compound 30: mp 209-210 °C; 1H NMR (400 MHz, CDCl3) δ 1.40 (t, J = 7.0 Hz, 3H, CH2CH3), 2.30-2.50 (m, 2H, CH2), 3.25-3.40 (m, 4H, piperazine CH2), 3.60-3.80 (m, 6H, piperazine CH2 and CH2), 4.30-4.50 (m, 4H, CH2CH3 and CH2), 6.60-6.75 (m, 2H, pyridine CH), 7.00 (s, 1H, H-8), 7.50 (t, J = 6.4 Hz, 1H, pyridine CH), 8.15-8.25 (m, 1H, pyridine CH), 8.40 (s, 1H, H-5), 8.80 (s, 1H, H-2).
2.1.22. Ethyl 6- Nitro -4-Oxo-1- [3-(Pyridin-2-Ylamino) Propyl]-7- (4-(Pyridin2-yl) Piperazin-1-yl]) -1,4- Dihydroquinoline -3- Carboxylate (31)
The title compound was obtained starting from 27 and 1-(2-pyridinyl)piperazine using Method A (80 °C, 3 h), in 100% yield: mp 124-125 °C; 1H NMR (400 MHz, CDCl3) δ 1.40 (t, J = 7.1 Hz, 3H, CH2CH3), 2.20-2.30 (m, 2H, NCH2CH2CH2NH), 3.10-3.20 (m, 4H, piperazine CH2), 3.50-3.60 (m, 2H, NCH2CH2CH2NH), 3.65-3.75 (m, 4H, piperazine CH2), 4.25-4.40 (m, 4H, NCH2CH2CH2NH and CH2CH3), 4.75-4.85 (m, 1H, NH), 6.35-6.45 (m, 1H, pyridine CH), 6.55-6.70 (m, 3H, pyridine CH), 6.80 (s, 1H, H-8), 7.30-7.40, 7.45-7.55, 8.05-8.15, and 8.15-8.25 (m, each 1H, pyridine CH), 8.45 (s, 1H, H-5), 8.85 (s, 1H, H-2).
2.1.23. Ethyl 6-Nitro-4-Oxo-1-(3-(Piperazin-1-Yl)Propyl)-7-(4-(Pyridin2-yl)Piperazin-1-yl])-1,4-Dihydroquinoline-3-Carboxylate (32)
Piperazine (0.08 g, 9.39 mmol) and DIPEA (0.12 g, 9.38 mmol) were added to a solution of 30 (0.13 g, 2.34 mmol) in dry DMF (5 mL), and the mixture was stirred at 80 °C for 48 h. After cooling, the reaction mixture was evaporated to dryness and the residue was purified by flash chromatography eluting with CHCl3/MeOH/NH4OH (from 90:10:0 to 79:20:1), to give 32 (0.08 g, 63%): mp 186-189 °C; 1H NMR (400 MHz, DMSO-d6) δ 1.15 (t, J = 7.0 Hz, 3H, CH2CH3), 1.90-2.00 and 2.10-2.15 (m, each 2H, CH2), 2.20-2.30, 2.90-3.0, 3.10-3.20 and 3.70-3.80 (m, each 4H, piperazine CH2), 4.10 (q, J = 7.0 Hz, 2H, CH2CH3), 4.20-4.30 (m, 2H, CH2), 6.70 (t, J = 6.5 Hz, 1H, pyridine CH), 6.90 (d, J = 9.3 Hz, 1H, pyridine CH), 7.10 (s, 1H, H-8), 7.60 (t, J = 6.5 Hz, 1H, pyridine CH), 8.10-8.20 (m, 1H, pyridine CH), 8.60 (s, 1H, H-5), 8.70 (s, 1H, H-2).
2.1.24. Ethyl 6-Amino-4-Oxo-1-(Pyridin-2-Ylmethyl)-7-(4-(Pyridin2-yl)Piperazin-1-yl])-1,4-Dihydroquinoline-3-Carboxylate (33)
The title compound was obtained starting from 28 using Method B (45 min) in 34% yield: mp 268-270 °C; 1H NMR (400 MHz, DMSO-d6) δ 1.30 (t, J = 7.0 Hz, 3H, CH2CH3), 2.75-2.85 and 3.60-3.70 (m, each 4H, piperazine CH2), 4.25 (q, J = 7.0 Hz, 2H, CH2CH3), 5.20 (bs, 2H, NH2), 5.70 (s, 2H, CH2), 6.70 (t, J = 5.0 Hz, 1H, pyridine CH), 6.85 (d, J = 8.5 Hz, 1H, pyridine CH), 7.00 (s, 1H, H-8), 7.30 (t, J = 5.2 Hz, 1H, pyridine CH), 7.35-7.45 (m, 2H, H-5 and pyridine CH), 7.60 and 7.80 (t, J = 7.0 Hz, each 1H, pyridine CH), 8.20 and 8.45 (d, J = 3.7 Hz, each 1H, pyridine CH), 8.75 (s, 1H, H-2).
2.1.25. Ethyl 6-Amino-4-Oxo-7-(4-(Pyridin2-yl)Piperazin-1-yl])-1-[3-(4-(Pyridin2-yl)Piperazin-1-yl])Propyl]-1,4-Dihydroquinoline-3-Carboxylate (34)
The title compound was obtained starting from 29 using Method B (1 h) in 88% yield: mp 233-234 °C; 1H NMR (400 MHz, DMSO-d6) δ 1.25 (t, J = 7.1 Hz, 3H, CH2CH3), 1.90-2.00 (m, 2H, CH2), 2.20-2.40 (m, 6H, CH2 and piperazine CH2), 3.10-3.20 and 3.40-3.50 (m, each 4H, piperazine CH2), 3.70-3.75 (m, 4H, piperazine CH2), 4.20 (q, J = 7.1 Hz, 2H, CH2CH3), 4.45-4.50 (m, 2H, CH2), 5.25 (bs, 2H, NH2), 6.65-6.70 (m, 2H, pyridine CH), 6.80 and 6.90 (d, J = 5.0 Hz, each 1H, pyridine CH), 7.15 (s, 1H, H-8), 7.45-7.55 (m, 3H, H-5 and pyridine CH), 8.10-8.20 (m, 2H, pyridine CH), 8.50 (s, 1H, H-2).
2.1.26. Ethyl 6 -Amino -4- Oxo-1- [3-(Pyridin-2-Ylamino) Propyl] -7- (4-(Pyridin2-yl) Piperazin-1-yl]) -1,4- Dihydroquinoline-3- Carboxylate (35)
The title compound was obtained starting from 31 using Method B (15 min) in 46% yield: mp 244-245 °C; 1H NMR (400 MHz, DMSO-d6) δ 1.25 (t, J = 7.1 Hz, 3H, CH2CH3), 1.95-2.05 (m, 2H, NCH2CH2CH2NH), 2.80-2.95 (m, 4H, piperazine CH2), 3.20-3.30 (m, 2H, NCH2CH2CH2NH), 4.60-4.75 (m, 4H, piperazine CH2), 4.20 (q, J = 7.1 Hz, 2H, CH2CH3), 4.25-4.35 (m, 2H, NCH2CH2CH2NH), 5.25 (s, 2H, NH2), 6.40-6.50 and 6.65-6.75 (m, each 2H, pyridine CH), 7.00 (s, 1H, H-8), 7.25-7.35 (m, 1H, pyridine CH), 7.50-7.60 (m, 2H, pyridine CH and H-5), 7.85-7.95 and 8.10-8.20 (m, each 1H, pyridine CH), 8.50 (s, 1H, H-2).
2.1.27. Ethyl 6-Amino-4-Oxo-1-(3-(Piperazin-1-Yl)Propyl)-7-(4-(Pyridin2-yl)Piperazin-1-yl])-1,4-Dihydroquinoline-3-Carboxylate (36)
The title compound was obtained starting from 32 using Method B (40 min) in 88% yield: mp 256-257 °C; 1H NMR (200 MHz, methanol-d4) δ 1.35 (t, J = 7.0 Hz, 3H, CH2CH3), 2.05-2.10 (m, 2H, CH2), 2.40-2.50 (m, 6H, CH2 and piperazine CH2), 2.90-3.00, 3.20-3.30, and 3.70-3.80 (m, each 4H, piperazine CH2), 4.35 (q, J = 7.0 Hz, 2H, CH2CH3), 4.60-4.65 (m, 2H, CH2), 6.70 (t, J = 6.5 Hz, 1H, pyridine CH), 6.90 (d, J = 9.3 Hz, 1H, pyridine CH), 7.20 (s, 1H, H-8), 7.60 (t, J = 6.5 Hz, 1H, pyridine CH), 7.65 (s, 1H, H-5), 8.10-8.20 (m, 1H, pyridine CH), 8.70 (s, 1H, H-2).
2.1.28. 6- Amino-4-Oxo-1- (Pyridin-2-Ylmethyl) -7-(4- (Pyridin2-yl) Piperazin-1-yl]) -1,4- Dihydroquinoline-3- Carboxylic Acid (4)
The title compound was obtained starting from 33 using Method C (20 h) after purification by treatment with Et2O in 64% yield: mp 287-288 °C; 1H NMR (400 MHz, DMSO-d6) δ 2.85-2.95 and 3.65-3.75 (m, each 4H, piperazine CH2), 5.45 (bs, 2H, NH2), 5.90 (s, 2H, CH2), 6.65 (t, J = 5.7 Hz, 1H, pyridine CH), 6.95 (d, J = 8.6 Hz, 1H, pyridine CH), 7.20 (s, 1H, H-8), 7.25-7.35 (m, 1H, pyridine CH), 7.50-7.60 (m, 3H, H-5 and pyridine CH), 7.80 (t, J = 7.1 Hz, 1H, pyridine CH), 8.20 and 8.50 (d, J = 3.3 Hz, each 1H, pyridine CH), 9.00 (s, 1H, H-2), 15.90 (s, 1H, COOH); 13C NMR (101 MHz, DMSO-d6) δ 45.08 (2C), 49.90 (2C), 58.54, 106.46, 106.88, 107.66, 108.38, 113.67, 122.78, 122.85, 123.78, 132.67, 137.85, 138.10, 142.33, 145.67, 147.55, 148.08, 150.01, 155.17, 159.43, 167.47, 176.88. Anal. calcd. for C25H24N6O3: C 65.78, H 5.30, N 18.41, found: C 70.03, H 5.32, N 18.48.
2.1.29. 6-Amino-4-Oxo-7-(4-(Pyridin2-yl)Piperazin-1-yl])-1-[3-(4-(Pyridin2-yl)Piperazin-1-yl])Propyl]-1,4-Dihydroquinoline-3-Carboxylic Acid (6)
The title compound was obtained starting from 34 using Method C (6 h) after purification by crystallization from DMF in 20% yield: mp > 300 °C; 1H NMR (400 MHz, DMSO-d6) δ 2.15-2.25 and 3.0-3.15 (m, each 2H, CH2), 3.25-3.70 (m, 10H, piperazine CH2), 3.75-4.00 (m, 4H, piperazine CH2), 4.25-4.45 and 4.65-4.80 (m, each 2H, piperazine CH2 and CH2), 5.50 (bs, 2H, NH2), 6.75 and 6.95 (t, J = 6.4 Hz, each 1H, pyridine CH), 7.10 (d, J = 9.0 Hz, 1H, pyridine CH), 7.30 (s, 1H, H-8), 7.40 (d, J = 9.0 Hz, 1H, pyridine CH), 7.60 (s, 1H, H-5), 7.75 and 7.95 (t, J = 6.4 Hz, each 1H, pyridine CH), 8.05-8.15 (m, 2H, pyridine CH), 8.80 (s, 1H, H-2), 11.60 (bs, 1H, COOH); 13C NMR (101 MHz, DMSO-d6) δ 23.52, 42.75, 46.51 (2C), 49.42 (2C), 50.68 (2C), 51.34, 53.00 (2C), 106.67, 107.44 (2C), 108.13, 113.39, 114.49 (2C), 123.15, 132.23, 135.9, 137.9, 142.31, 145.50, 146.31 (2C), 151.06, 152.93, 153.60, 167.33, 176.74. Anal. calcd. for C31H36N8O3: C 65.47, H 6.38, N 19.70, found: C 65.40, H 6.70, N 19.55.
2.1.30. 6-Amino-4-Oxo-1-[3-(Pyridin-2-Ylamino)Propyl]-7-(4-(Pyridin2-yl)Piperazin-1-yl])-1,4-Dihydroquinoline-3-Carboxylic Acid Hydrochloride (7)
The title compound was obtained starting from 35 using Method C (6 h) after purification by treatment with Et2O in 91% yield: mp 290 °C (c); 1H NMR (400 MHz, DMSO-d6) δ 2.00-2.10 (m, 2H, CH2CH2CH2), 3.10-3.20 (m, 4H, piperazine CH2), 3.40-3.50 (m, 2H, NCH2), 3.90-4.00 (m, 4H, piperazine CH2), 4.65-4.75 (m, 2H, CH2NH), 6.75-6.85 and 6.90-7.00 (m, each 1H, pyridine CH), 7.10-7.20 (m, 1H, pyridine CH), 7.35-7.45 (m, 2H, H-8 and pyridine CH), 7.60 (s, 1H, H-5), 7.80-8.00 (m, 3H, pyridine CH), 8.10-8.20 (m, 1H, pyridine CH), 8.75 (s, 1H, H-2), 9.35 (s, 1H, NH); 13C NMR (101 MHz, DMSO-d6) δ 28.42, 39.45, 46.81 (2C), 49.62 (2C), 51.63, 106.79, 108.62, 112.39 (2C), 112.98, 113.36 (2C), 123.10, 133.11, 135.91, 137.90, 140.91, 144.44 (2C), 146.00, 146.55, 152.45, 152.96, 167.20, 176.64. Anal. calcd. for C27H30ClN7O3: C 60.50, H 5.64, N 18.29, found: C 60.62, H 5.77, N 18.33.
2.1.31. 6-Amino-4-Oxo-1-(3-(Piperazin-1-Yl)Propyl)-7-(4-(Pyridin2-yl)Piperazin-1-yl])-1,4-Dihydroquinoline-3-Carboxylic Acid (8)
The title compound was obtained starting from 36 using Method C (4 h) after purification by crystallization from DMF in 60% yield: mp > 300 °C; 1H NMR (400 MHz, DMSO-d6) δ 2.00-2.20 (m, 2H, CH2), 3.00-3.25 and 3.30-3.55 (m, each 8H, piperazine CH2), 3.90-4.00 and 4.50-4.60 (m, each 2H, CH2), 6.90 (t, J = 6.5 Hz, 1H, pyridine CH), 7.20 (s, 1H, H-8), 7.50 (d, J = 9.1 Hz, 1H, pyridine CH), 7.70 (s, 1H, H-5), 7.90-8.00 (m, 2H, pyridine CH), 8.70 (s, 1H, H-2); 13C NMR (101 MHz, DMSO-d6) δ 23.48, 42.69, 45.32 (2C), 56.12 (2C), 50.73 (2C), 51.69, 53.25 (2C), 106.18, 107.35, 108.23, 113.45, 114.61, 123.38, 132.91, 136.12, 142.75, 145.56, 146.42, 151.18, 153.06, 167.45, 176.88. Anal. calcd. for C26H33N7O3: C 63.52, H 6.77, N 19.95, found: C 63.79, H 7.02, N 20.13.
2.1.32. Ethyl 7- Chloro-4-Oxo-1- [2-(4-(Pyridin2-yl) Piperazin-1-yl]) Ethyl] -1,4-Dihydro-1,8- Naphthyridine-3- Carboxylate (40)
The title compound was obtained starting from 37 [32] and [2-(4-(Pyridin2-yl)Piperazin-1-yl])ethyl]amine [33] using Method D (2 h) to give intermediate 38, followed by Method E in 56% overall yield: mp 123-124° C; 1H NMR (400 MHz, DMSO-d6) δ 1.30 (t, J = 7.0 Hz, 3H, CH2CH3), 2.45-2.55 (m, 4H, piperazine CH2), 2.75 (t, J = 6.7 Hz, 2H, CH2), 3.40-3.50 (m, 4H, piperazine CH2), 4.20 (q, J = 7.0 Hz, 2H, CH2CH3), 4.60 (t, J = 6.7 Hz, 2H, CH2), 6.60-6.65 (m, 1H, pyridine CH), 6.80 (d, J = 8.7 Hz, 1H, pyridine CH), 7.50-7.60 (m, 1H, pyridine CH), 7.65 (d, J = 8.3 Hz, 1H, H-6), 8.10-8.15 (m, 1H, pyridine CH), 8.60 (d, J = 8.3 Hz, 1H, H-5), 8.85 (s, 1H, H-2).
2.1.33. Ethyl 7- Chloro-4-Oxo-1- [3-(4-(Pyridin2-yl) Piperazin-1-yl]) Propyl]-1,4- Dihydro-1,8- Naphthyridine-3- Carboxylate (41)
The title compound was obtained starting from 37 [32] and [3-(4-(pyridin-2-yl)piperazin-1-yl)propyl]amine [33] using Method D (2 h) to give intermediate 39, followed by Method E in 59% overall yield: mp 124-125 °C; 1H NMR (400 MHz, DMSO-d6) δ 1.30 (t, J = 7.0 Hz, 3H, CH2CH3), 2.00 (t, J = 6.7 Hz, 2H, NCH2CH2), 2.40-2.50 (m, 6H, CH2 and piperazine CH2) 3.40-3.50 (m, 4H, piperazine CH2), 4.25 (q, J = 7.0 Hz, 2H, CH2CH3), 4.50 (t, J = 6.7 Hz, 2H, NCH2CH2), 6.60-6.65 (m, 1H, pyridine CH), 6.80 (d, J = 8.7 Hz, 1H, pyridine CH), 7.50-7.60 (m, 1H, pyridine CH), 7.65 (d, J = 8.3 Hz, 1H, H-6), 8.10-8.15 (m, 1H, pyridine CH), 8.55 (d, J = 8.3 Hz, 1H, H-5), 8.90 (s, 1H, H-2).
2.1.34. Ethyl 7-[4-(1,3-Benzothiazol-2-Yl)Piperazin-1-Yl]-4-Oxo-1-[2-(4-(Pyridin2-yl)Piperazin-1-yl])Ethyl]-1,4-Dihydro-1,8-Naphthyridine-3-Carboxylate (42)
The title compound was obtained starting from 40 and 1-(1,3-benzothiazol-2-yl)piperazine [34] using Method A (80 °C, 24 h). After cooling the reaction mixture was poured into ice/water and extracted with CH2Cl2. The organic layers were evaporated to dryness affording an oil, which was tritured with Et2O, giving a solid which was filtered, to afford 42 in 68% yield: mp 124-125 °C; 1H NMR (400 MHz, DMSO-d6) δ 1.25 (t, J = 7.0 Hz, 3H, CH2CH3), 2.50-2.60 (m, 4H, piperazine CH2), 2.75 (t, J = 6.7 Hz, 2H, CH2), 3.40-3.50, 3.65-3.75, and 3.80-3.90 (m, each 4H, piperazine CH2), 4.20 (q, J = 7.0 Hz, 2H, CH2CH3), 4.75 (t, J = 6.7 Hz, 2H, CH2), 6.60-6.65 (m, 1H, pyridine CH), 6.80 (d, J = 8.7 Hz, 1H, pyridine CH), 7.00-7.10 (m, 2H, H-6 and benzothiazole CH), 7.25 (t, J = 7.4 Hz, 1H, benzothiazole CH), 7.40-7.50 (m, 2H, pyridine CH and benzothiazole CH), 7.75 (d, J = 7.9 Hz, 1H, benzothiazole CH), 8.05-8.10 (m, 1H, pyridine CH), 8.20 (d, J = 8.8 Hz, H-5), 8.60 (s, 1H, H-2).
2.1.35. Ethyl 7-[4-(1,3-Benzothiazol-2-Yl)Piperazin-1-Yl]-4-Oxo-1-[3-(4-(Pyridin2-yl)Piperazin-1-yl])Propyl]-1,4-Dihydro-1,8-Naphthyridine-3-Carboxylate (43)
The title compound was obtained starting from 41 and 1-(1,3-benzothiazol-2-yl)piperazine [34] using Method A (80 °C, 24 h) in 63% yield: mp 140-141 °C; 1H NMR (400 MHz, DMSO- d6) δ 1.30 (t, J = 7.0 Hz, 3H, CH2CH3), 2.00 (t, J = 6.7 Hz, 2H, NCH2CH2), 2.40-2.50 (m, 6H, CH2 and piperazine CH2), 3.40-3.50, 3.65-3.75 and 3.85-3.95 (m, each 4H, piperazine CH2), 4.20 (q, J = 7.0 Hz, 2H, CH2CH3), 4.40 (t, J = 6.7 Hz, 2H, NCH2CH2), 6.60-6.65 (m, 1H, pyridine CH), 6.80 (d, J = 8.7 Hz, 1H, pyridine CH), 7.00-7.10 (m, 2H, H-6 and benzothiazole CH), 7.25 (t, J = 7.4 Hz, 1H, benzothiazole CH), 7.40-7.50 (m, 2H, pyridine CH and benzothiazole CH), 7.75 (d, J = 7.9 Hz, 1H, benzothiazole CH), 8.05-8.10 (m, 1H, pyridine CH), 8.20 (d, J = 8.8 Hz, H-5), 8.60 (s, 1H, H-2).
2.1.36. 7-[4-(1,3-Benzothiazol-2-Yl)Piperazin-1-Yl]-4-Oxo-1-[2-(4-(Pyridin-2-Yl)Piperazin-1-Yl)Ethyl]-1,4-Dihydro-1,8-Naphthyridine-3-Carboxylic Acid (11)
The title compound was obtained starting from 42 using Method C (2 h) after purification by crystallization from DMF in 41% yield: mp 294-295 °C (dec.) °C; 1H NMR (400 MHz, DMSO-d6) δ 2.50-2.60 (m, 4H, piperazine CH2), 2.75 (t, J = 6.7 Hz, 2H, CH2), 3.30-3.40, 3.65-3.75, and 3.90-4.00 (m, each 4H, piperazine CH2), 4.65 (t, J = 6.7 Hz, 2H, CH2), 6.60-6.65 (m, 1H, pyridine CH), 6.80 (d, J = 8.7 Hz, 1H, pyridine CH), 7.05 (t, J = 7.7 Hz, 1H, benzothiazole CH), 7.20-7.30 (m, 2H, H-6 and benzothiazole CH), 7.40-7.50 (m, 2H, pyridine CH and benzothiazole CH), 7.75 (d, J = 7.9 Hz, 1H, benzothiazole CH), 8.05-8.10 (m, 1H, pyridine CH), 8.30 (d, J = 8.8 Hz, H-5), 8.80 (s, 1H, H-2), 15.50 (s, 1H, COOH); 13C NMR (101 MHz, DMSO-d6) δ 44.21 (2C), 45.27, 47.86 (4C), 52.82 (2C), 56.23, 107.61, 107.81, 108.41, 111.50, 113.54, 119.20, 121.75, 121.88, 126.53, 130.90, 136.42, 137.98, 147.99, 149.62, 150.01, 152.80, 159.29, 159.46, 166.63, 168.47, 177.04. Anal. calcd. for C31H32N8O3S: C 62.40, H 5.41, N 18.78, found: C 62.25, H 5.70, N 18.65.
2.1.37. 7-[4-(1,3-Benzothiazol-2-Yl)Piperazin-1-Yl]-4-Oxo-1-[3-(4-(Pyridin-2-Yl)Piperazin-1-Yl)Propyl]-1,4-Dihydro-1,8-Naphthyridine-3-Carboxylic Acid (12)
The title compound was obtained starting from 43 using Method C (2 h) after purification by crystallization from DMF in 16% yield: mp 256-257 °C (dec); 1H NMR (400 MHz, DMSO-d6) δ 2.00 (t, J = 6.7 Hz, 2H, CH2), 2.25-2.35 (m, 6H, CH2 and piperazine CH2), 3.25-3.35, 3.65-3.75 and 3.95-4.00 (m, each 4H, piperazine CH2), 4.50 (t, J = 6.7 Hz, 2H, CH2), 6.60-6.65 (m, 1H, pyridine CH), 6.75 (d, J = 8.7 Hz, 1H, pyridine CH), 7.05 (t, J = 7.7 Hz, 1H, benzothiazole CH), 7.20-7.30 (m, 2H, H-6 and benzothiazole CH), 7.40-7.50 (m, 2H, pyridine CH and benzothiazole CH), 7.75 (d, J = 7.9 Hz, 1H, benzothiazole CH), 8.05-8.10 (m, 1H, pyridine CH), 8.30 (d, J = 8.8 Hz, H-5), 9.00 (s, 1H, H-2), 15.50 (s, 1H, COOH); 13C NMR (101 MHz, DMSO-d6) δ 23.62, 42.93, 46.48 (2C), 48.31 (2C), 49.16 (2C), 51.42, 52.87 (2C), 107.53, 108.16, 108.43, 111.62, 113.48, 119.1, 122.15, 122.88, 126.41, 131.10, 136.72, 138.22, 148.34, 150.21, 150.34, 152.98, 159.75, 159.88, 166.89, 169.17, 177.25. Anal. calcd. for C32H34N8O3S: C 62.93, H 5.61, N 18.35, found: C 62.98, H 5.60, N 18.35. .
2.2. Biology
2.2.1. In vitro Anti-HIV Assays
The evaluation of the antiviral activity of the target compounds against HIV-1 strain IIIB in MT-4 cells was performed using the MTT assay as previously described [35, 36]. Mock-infected cells were used to assess the cytotoxic effects of the test compounds.
2.2.2. Fluorescence Quenching Assay (FQA)
The effect of tested quinolones on the Tat-TAR complex was evaluated using an FQA, a FRET-based competition assay, as previously described [23], using the Tat-derived peptide labelled with the donor fluorescein at its N terminus and the 29 nt wtTAR labeled at its 3’-end with a dabcyl moiety (quencher).
3. RESULTS AND DISCUSSION
3.1. Chemistry
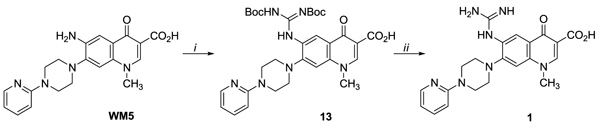
The introduction of the guanidine group at C-6 position in the quinolone derivative 1 was accomplished by reacting WM5 [20] with N,N′-di-(tert-butoxycarbonyl)thiourea in the presence of HgCl2 followed by Boc deprotection of intermediate 13 under acid conditions Scheme (1).
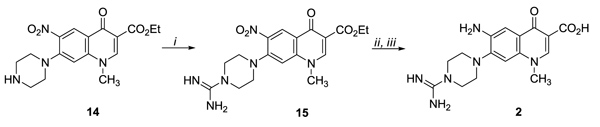
The synthesis of the C-7 4-carbamimidoylpiperazine derivative 2 started by reacting derivative 14 [28] with methyl imidothiocarbamate sulfate to give intermediate 15, which, after reduction of the nitro group accomplished with FeSO4 and NH4OH followed by acid hydrolysis of the ethyl ester intermediate, led to the target compound 2 Scheme (2).
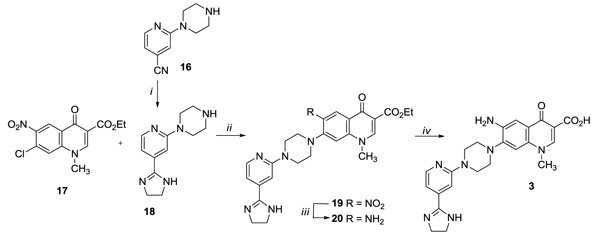
The C-7 imidazolinpyridinpiperazine derivative 3 was synthesized, as outlined in Scheme (3), by reacting intermediate 17 [20] with the 1- [4- (4,5-dihydro-1H-imidazol-2-yl) pyridin-2-yl] piperazine 18 in DMF, affording intermediate 19. Nitro derivative 19 was then catalytically reduced to amino derivative 20 and finally hydrolysed under basic conditions to give the target compound 3. The required building block 18 was in turn prepared through the reaction of nitrile derivative 16 [29] with ethylenediamine in presence of p-toluenesulfonic acid.
The synthesis of quinolone derivatives 4 and 6-8 Scheme (4) was accomplished through a cycloaracylation procedure. Thus, acrylate 21 [30] was reacted with (pyridin-2-ylmethyl) amine, (3-bromopropyl) amine, and N-(pyridin-2-yl) propane-1,3-diamine in Et2O/EtOH mixture at room temperature to give intermediates 22-24, which by treatment with K2CO3 in dry DMF at 80 °C afforded synthons 25-27. These derivatives were then subjected to nucleophilic reaction with 1-(2-pyridinyl) piperazine in dry DMF, to give intermediates 28-31. In the case of compound 26, the nucleophilic reaction led to the mixture of the di-substituted 29 and mono-substituted 30, which were easily isolated by chromatographical purification. Intermediate 30 was then functionalized at the N-1 position by reaction with piperazine in presence of DIPEA in dry DMF affording derivative 32. Then, the 6-nitro-derivatives 28, 29, 31, and 32 were catalytically reduced to give 6-amino-derivatives 33-36. Finally, the target compounds 4 and 6-8 were obtained by saponification of the corresponding ethyl esters 33-36.
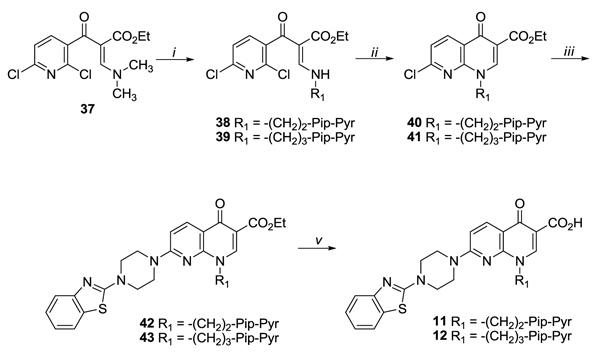
By following an analogous synthetic route, 1,8-naphthyridone derivatives 11 and 12 were prepared as presented in Scheme (5). Thus, the reaction of acrylate 37 [32] with [2-(4-(pyridin-2-yl)piperazin-1-yl)ethyl]amine [33] and [3-(4-(pyridin-2-yl)piperazin-1-yl)propyl]amine [33] in Et2 O/ EtOH mixture at room temperature gave intermediates 38 and 39, which by treatment with K2CO3 in dry DMF at 80 °C, afforded synthons 40 and 41, respectively. The nucleophilic reaction of naphthyridone derivatives 40 and 41 with 1- (1,3- benzothiazol-2-yl) piperazine [34] led to intermediates 42 and 43. Finally, the target compounds 11 and 12 were obtained by saponification of the corresponding ethyl esters 42 and 43.

| Compd |
EC50 (µM)a,c HIV-1 (IIIB) |
CC50 (µM)b,c | SId | Ki Tat-TAR (µM)e |
|---|---|---|---|---|
| 1 | >296 | >296 | 1 | N.T. f |
| 2 | >363 | 363 | 1 | N.T. |
| 3 | >34.9 | 34.9 ± 1.43 | <1 | N.T. |
| 4 | >19.8 | 19.8 ± 5.02 | <1 | N.T. |
| 5g | 5.05 ± 0.41 | 20.9 ± 3.93 | 4 | 1.15 ± 0.55 |
| 6 | 1.21 ± 0.08 | 5.92 ± 2.19 | 5 | 0.91 ± 0.27 |
| 7 | ≥3.42 | 3.94 ± 0.62 | ≤1 | 2.18 ± 0.40 |
| 8 | 70.8 ± 0.71 | >254 | >4 | N.T. |
| 9g | 1.16 ± 0.18 | 3.97 ± 0.70 | 3 | N.D.h |
| 10g | ≥0.18 | 3.34 ± 1.47 | ≤18 | 1.72 ± 0.22 |
| 11 | 3.37 ± 1.62 | 11.65 ± 1.17 | 3.5 | N.D. |
| 12 | ≥0.67 | 10.70± 7.76 | ≤16 | N.D. |
| WM5i | 0.15 ± 0.05 | 2.21 ± 1.05 | 15 | 2.22 ± 0.66 |
3.2. Biological Evaluation
The newly synthesized compounds were initially evaluated for anti-HIV-1 (IIIB) activity in MT-4 cells, determining their cytotoxicity in parallel Table 1. WM5 [20] was evaluated in the same cell lines for comparative purposes.
From the results, it emerged that the introduction of a C-6 guanidine group (compound 1) as well as an amidine moiety and a 4-imidazolinpyridine ring at the C-7 position (compounds 2 and 3) was completely detrimental in the inhibition of HIV replication. However, the low cytotoxicity showed by the compounds could suggest their inability to enter the cells.
The introduction of a 2-pyridinylmethyl moiety at N-1 position (compound 4) did not produce an active derivative (EC50 > 19.8 µM). Conversely, the presence of a (pyridin-2-yl) piperazine spaced by an ethylene or propylene unit permitted to restore the anti-HIV-1 activity in compounds 5 and 6 (EC50 = 5.05 and 1.21 µM, respectively) at no cytotoxic concentrations.
By alternatively deleting the piperazine or pyridine ring in compound 6, no activity at subcytotoxic concentrations was observed for compound 7, while only a weak inhibitory activity was conserved by compound 8. More interesting results were achieved when protonable side chains were inserted at the N-1 position of the 1,8-naphthyridone scaffold. In particular, the introduction of the (pyridin-2-yl)piperazine spaced by an ethylene or propylene unit gave compounds 11 and 12, both endowed with good anti-HIV-1 activity (EC50 = 3.37 and ≥0.67 µM) coupled with SI values of 3.5 and ≤16, respectively. The deletion of the piperazine ring, as in compound 9 and 10, also in this case permitted to maintain anti-HIV-1 (EC50 = 1.16 and ≥0.18 µM, SI = 3 and ≤18, respectively), with compound 10 that emerged as the most potent of the series.
In order to investigate the ability of active anti-HIV-1 compounds (derivative 5-7 and 9-12) to interfere with the formation of the Tat-TAR complex, they were evaluated in a FQA using a truncated Tat peptide. Unfortunately, solubility issues did not allow the evaluation of compounds 9, 11 and 12. Derivatives 5, 6, and 10 appeared to be able to inhibit the Tat-TAR complex formation, all presenting Ki values even improved as compared to the values obtained with WM5 Table 1. In particular, enhancing the distance of the pyridine ring from the quinolone nucleus, the ability of the compounds to inhibit the formation of the complex peptide-TAR increases: Ki 6 < 5 < 7; moreover, the presence of a piperazine in the side chain (compounds 6 and 5) has a positive effect on the activity of compounds, probably because it confers more rigidity to the chain and the position of the pyridine ring respect to the quinolone nucleus is fixed. Even if a strict correlation between the Ki values and the anti-HIV activities did not exist, this assay confirmed that the impairment of the Tat-TAR complex formation represents a putative target for the new derivatives.
CONCLUSION
The Tat-mediated transcription, the only phase of the HIV-1 replicative cycle in which viral genome amplification occurs, is a really attractive step that if inhibited by a suitable inhibitor could lead to a functional cure or even HIV infection eradication. For years, the inhibition of the Tat-mediated transcription has been highly pursued through different approaches, but this step of the viral replicative cycle remains untouched by any of the drugs in therapy. However, there are examples of promising Tat-mediated transcription inhibitors worthy to be mentioned, such as triptolide [37] and dCA [38] working on Tat protein.
In the attempt to obtain potent TAR binders, we started from WM5, a small molecule endowed with potent anti-HIV and Tat-mediated transcription inhibition activity thanks to the ability to selectively bind the bulge region of TAR; indeed no interaction with TAR-unrelated nucleic acids such as tRNA sequence and single-stranded or double-stranded DNA structures was observed [39].
Starting from WM5, various protonable moieties were inserted in different positions of the quinolone scaffold, intended to mime the basic region of Tat involved in the TAR interaction. The modifications introduced in the structure do not result in loss of activity, but rather seem to improve the activity as inhibitors of the Tat-TAR complex formation, leading to the identification of compounds able to recognize TAR better than WM5. Important SAR insights were also achieved for the quinolone class of anti-HIV derivatives. In particular, the C-6 and C-7 positions were confirmed as particularly sensitive to structural modifications, while the N-1 emerged as a suitable position to host protonable moieties such as the pyridine-based side chains. This is an important new insight since the most potent quinolones reported until now were all characterized by the presence of small alkyl groups at the N-1 position [22, 40]. In addition, potent anti-HIV compounds were identified, with naphthyridone derivatives 10 and 12 which showed EC50 values in the sub-micromolar range.
In conclusion, the addition of protonable chains as tool to improve the TAR binding properties permitted to obtain active compounds, but additional knowledge on the interaction of TAR RNA with viral protein Tat and host Super Elongation Complex (SEC) [41, 42] are still necessary to design compounds able to better recognize the TAR RNA and potently inhibit HIV replication. During the reviewing of this work, two interesting papers were published that could help the design of an effective TAR binder. In particular, Schulze-Gahmen and Hurley solved the crystal structure of the TAR in complex with Tat and the SEC core (CycT1/CDK9/AFF4) [43], while Varani and co-workers reported the NMR structure of TAR RNA in complex with a ultra-potent macrocyclic peptide that mimics the Arginine Rich Motif (ARM) of Tat [44], which however only weakly inhibit the Tat-mediated transcription.
These studies revealed how the direct interaction of Tat-ARM with TAR RNA bulge is required to induce the RNA structural change, but it is the recognition of the loop by the CycT1 that dominantly contributes to the binding energy. Thus, a future TAR binder should be able to target not only the UCU bulge but above all the interaction of CycT1 with the rearranged TAR RNA loop, to achieve an effective Tat-mediated transcription inhibition in cells.
LIST OF ABBREVIATIONS
| ARM | = Arginine Rich Motif |
| SAR | = Structure-Activity Relationship |
| SEC | = Super Elongation Complex |
| TAR | = Transactivation Response Element |
| FQA | = Fluorescence Quenching Assay. |
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable
HUMAN AND ANIMAL RIGHTS
No Animals/Humans were used for studies that are basis of this research.
CONSENT FOR PUBLICATION
Not applicable
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial, or otherwise.
ACKNOWLEDGEMENTS
We thank K. Erven, K. Uyttersprot and C. Heens for technical assistance with the evaluation of the anti-HIV activity and the assessment of cytotoxicity.

