All published articles of this journal are available on ScienceDirect.

Evaluation of Scoring Function Performance on DNA-ligand Complexes
Abstract
Background:
DNA has been a pharmacological target for different types of treatment, such as antibiotics and chemotherapy agents, and is still a potential target in many drug discovery processes. However, most docking and scoring approaches were parameterised for protein-ligand interactions; their suitability for modelling DNA-ligand interactions is uncertain.
Objective:
This study investigated the performance of four scoring functions on DNA-ligand complexes.
Material & Methods:
Here, we explored the ability of four docking protocols and scoring functions to discriminate the native pose of 33 DNA-ligand complexes over a compiled set of 200 decoys for each DNA-ligand complexes. The four approaches were the AutoDock, ASP@GOLD, ChemScore@GOLD and GoldScore@GOLD.
Results:
Our results indicate that AutoDock performed the best when predicting binding mode and that ChemScore@GOLD achieved the best discriminative power. Rescoring of AutoDock-generated decoys with ChemScore@GOLD further enhanced their individual discriminative powers. All four approaches have no discriminative power in some DNA-ligand complexes, including both minor groove binders and intercalators.
Conclusion:
This study suggests that the evaluation for each DNA-ligand complex should be performed in order to obtain meaningful results for any drug discovery processes. Rescoring with different scoring functions can improve discriminative power.
1. INTRODUCTION
DNA is an important target for various types of treatments, most notably in cancer [1]. Certain chemotherapy agents, such as doxorubicin and daunorubicin are classic examples of small molecules that bind to double-stranded DNA and produce therapeutic effects [2, 3]. Recently, Zhu et al. [4] discovered that drug-DNA adducts may serve as clinical applications in targeting anticancer drug delivery to reduce the risks of adverse reactions caused by chemotherapies. Apart from the therapeutic effects, drug molecules with a high ability to bind DNA may cause toxicity, particularly genotoxicity. Classic examples are genistein, thalidomide and certain antihistamines [5, 6]. These certainly demonstrate the importance of studying the interactions between DNA and small ligand molecules. However, only a few computational studies were performed on DNA-ligand complexes and no related systematic performance assessments were performed with most of the computational modelling software [7-9].
Docking is a popular technique of computational drug design to study the interactions between drug molecules and their associated receptors [10]. It consists of two main components, which are docking algorithms and scoring functions. The former is responsible for generating binding modes and the latter calculate the binding affinity of each mode and ‘rank’ their orders. In the past two decades, many scoring functions have been developed, which are classified as physics-based methods, empirical scoring functions, knowledge-based potentials, and descriptor-based scoring functions [11]. Most were parameterised or calibrated to simulate protein-ligand interactions and their performance assessments were focused on protein-ligand complexes. Hence, their abilities to score DNA-ligand bonds are uncertain.
Small drug molecules generally bind to minor or major grooves of the DNA, while others bind between the base pairs of two DNA molecules, i.e. intercalators, to produce pharmacological effects. In comparison to the active sites of proteins, the DNA grooves are not as specific and well-defined for binding [12, 13]. Therefore, the simulation of DNA-ligand interactions is generally considered challenging.
Performance assessments of docking software have been reported in various studies [14]; however, they are mostly focused on protein-ligand docking. These studies pointed out that the accuracy of docking simulations is dependent on the type of algorithm, scoring functions and complexes [10]. In some cases, the accuracy of protein-ligand docking reached >90%, but 0% was also reported in the literature [10, 15]. Recent studies by Li et al. [14, 16] assessed the performance of 20 scoring functions among 195 protein-ligand complexes; the results revealed that ChemScore, ChemPLP and PLP are the top-ranked functions in the scoring, ranking and docking power tests. Regarding DNA-ligand docking, only a couple of accuracy assessments can be found in the literature, which were focused on one or two docking programmes. For instance, AutoDock [17] and Surflex [18] accurately reproduced crystallographic structures (<2.00Å root mean square deviation, RMSD) of intercalators, daunorubicin and the minor groove binders distamycin and pentamidine [7]. Another recent study analysed the ability of AutoDock to accurately dock ligands into 63 preformed DNA intercalation sites [9]. Their results indicated that AutoDock was able to generate conformations with RMSD values <2.00Å in approximately 80% of cases [9]. These studies successfully assessed the docking power and binding mode, but substantially missed that of the scoring and ranking power, i.e. the ability of a scoring function to produce binding scores that correlate well with experimental binding data. Therefore, a comprehensive study to assess docking and scoring abilities of various scoring functions is required to aid the selection of programmes to perform DNA-ligand docking simulations. Docking and scoring abilities generally indicate the accuracy of binding mode predictions and binding affinity predictions, i.e. the ability to discriminate between binders and non-binders.
Many studies assessing protein-ligand scoring functions have identified their advantages and disadvantages. Different types of scoring function require different parameterisations in different levels of accuracy and with different levels of quantity. In general, a large amount of binding affinity data from numerous type of complexes is required for an accurate universal application of empirical-based scoring functions [15]. Accurate inclusion of the desolvation, entropic effects and atom-type recognition are crucial for force field-based and knowledge-based functions [19]. Overall, most scoring functions rely on simple equations to calculate docking scores, and this simplicity of functions can certainly reduce the computational cost and allow the application of screening large databases [20]. However, these simple equations may lead to weak correlations with experimental data. Therefore, scientists have been using computationally expensive methods, such as molecular dynamics (MD), to validate or rescore their docking results. Most MDs are molecular mechanic (MM)-based force field methods, which generally neglect important explicit interaction energies, including charge transfer and electronic polarisation energies [21]. These pitfalls attracted the investigation of the combined quantum mechanical/molecular mechanical (QM/MM) scoring functions. These methods are not only computationally expensive but may require a substantial amount of time to set up the simulations. Hence, a highly accurate docking programme or scoring function may prevent the need to perform these computationally demanding processes. Studies have shown that using consensus scoring (CS), i.e. combining/rescoring scoring functions on protein-ligand docking have dramatically reduced false negatives and significantly enhanced hit-rates by compensating for the deficiencies of individual scoring function [22, 23].
This study evaluated the performance of four scoring functions on DNA-ligand docking. They were ChemScore [24], GoldScore [25], ASP (Astex Statistical Potential) [26] and AutoDock [27]. The former three were implemented in the GOLD v5.5 software [28], whereas the last was accessed via the AutoDock 4.2 docking programme [27]. Cross-rescoring were also performed to explore ways to improve the performance of the scoring functions.
2. MATERIALS AND METHODS
2.1. Selection of DNA Complexes
A search of the Protein Data Bank (www.rcsb.org) using the keyword ‘DNA-ligand’ was performed. A set of rules based on the study of Li et al. [16] on filtering high quality protein-ligand complexes was employed in this study to select the DNA-ligand complexes in our test set. They are: (1) The complex structures must be derived from crystallographic experiments and not NMR-resolved; (2) Resolution must be better than 2.50Å; (3) R-factor must be lower than 0.250; (4) DNA-protein complexes are excluded; (5) Complexes with low (Kd or Ki > 10 mM) binding affinities are excluded; (6) Estimated binding datae.g., Kd ~ 1 nM or Ki > 10 μM, are excluded.; (7) Ligands with cofactors or substrates located closely (within 5.00Å) are excluded; (8) Ligands must not be solvent or buffer components.
2.2. Decoy Generation
In order to evaluate the performance of the docking and scoring suites, geometric decoys were generated using the two docking programmes and their four associated scoring functions. These were the ChemScore@GOLD [24], Gold Score@GOLD [25], ASP@GOLD [26] and AutoDock scoring functions [27]. Geometric decoys are the crystallographic ligand embedded in the DNA-ligand complexes with different binding modes. Decoys have been widely used as a tool to evaluate and improve molecular docking scoring functions [29]. In this study, the decoys were basically the top 50 ranked binding modes of each ligand generated by each docking algorithm and its associated scoring function. In order to reduce bias, each of the four scoring functions was responsible for generating the same number of decoys (fifty), which were combined together to compile a set of 200 (4 x 50) decoys for each ligand.
2.3. Docking and Scoring Methods
In this study, two software programmes were employed for docking and scoring: GOLD v5.5 [28] and AutoDock 4.2 [27]. GOLD (Genetic Optimisation for Ligand Docking) is a widely used commercial programme of the CSD-Discovery and CSD-Enterprise Suites. AutoDock is freely available under the GNU General Public License (http://AutoDock.scripps.edu/). Both docking programmes adopt different scoring functions to optimise fitness scores using the evolutionary docking algorithm, which is also called genetic algorithm. The aim of a docking algorithm is to adjust the binding modes of a ligand within its receptor to its lowest possible energy state. Genetic algorithms work by randomly assigning a population of potential solutions initially. These solutions represent the potential docking modes of the ligand. Then, each docking mode of the population is encoded as a chromosome and each chromosome is assigned a fitness score calculated by a scoring function. The score represents the binding affinity and is used to rank the chromosomes within the population. The score is dependent on the ligand geometry parameters of the chromosomes, such as the mapping of H-bond atoms, Van der Waals interactions and hydrophobic points between the ligand and its receptor. The population of chromosomes is then repeatedly optimised through migration, mutation and crossover until the fittest member of the population is achieved, i.e. evolutionary cycles continue until a docking mode with best fitness score is obtained.
The accuracy of a docking programme is dependent on both the docking algorithms and the scoring functions. Therefore, although the docking algorithms between two programmes are similar, the simulated best fitness binding mode of the same ligand may be different. In this study, each docking programme and its associated scoring functions were employed to generate decoys, this helps to obtain a set of ligand binding modes that was as comprehensive as possible.
The initial set up of the molecular structures for all of the docking simulations of the two programmes was identical. The crystal structures of all of the selected DNA-ligand complexes were downloaded from the Protein Data Bank (https://www. rcsb. org). All ions, ligands and water molecules were then removed and hydrogen was added before the docking simulations. The docking programme GOLD was employed to perform docking on each DNA-ligand complex with the scoring functions ChemScore [24], GoldScore [25] and ASP [26] independently. Genetic algorithms with 100% search efficiency, no possibility of early termination, and a slow option were used. All parameters were set as defaults. Binding sites were defined as all atoms within 6Å of the cognate ligands in the crystal structures. The AutoDock docking procedures were composed of two steps: (1) using the Lamarkian Genetic Algorithm to sample the ligand conformation in the binding sites of selected DNA, based on the pre-calculated energy grids [30], where the binding site was defined as all atoms within 6Å of the cognate ligands, the grid spacing was set to 0.375Å, and the number of evaluations per docking run was 2,500,000; and (2) the AutoDock scoring function was subsequently used to determine the binding scores of the different conformations, which then served as geometric decoys.
2.4. Evaluation Methods
Many evaluation methods of scoring functions have been documented in literatures [10, 31, 32]. One of the most comprehensive studies recently evaluated 20 scoring functions on docking, scoring and ranking power of 195 protein-ligand complexes [14, 16]. Docking power was generally assessed by calculating the success rate of a scoring function by counting the number of best scored binding modes with an RMSD of less than 2.0Å to its native crystallographic binding mode. Scoring and ranking power may be assessed by comparing binding affinities and ligand ranking orders obtained from experimental data and docking simulations using the classic Pearson’s correlation coefficient. These common evaluation methods required experimental binding affinities that can generally be obtained through literature and database searches. However, finding the experimental binding affinities for all of the selected DNA-ligand complexes in this study was impossible, as most did not have a documented binding affinity. Moreover, some complexes have two or more different binding affinities, which caused confusion in choosing the binding affinity that should be used for evaluation. The different values of binding affinities of the same complex were probably due to the different experimental procedures/ environments in different laboratories. As the above reasons may introduce bias to our results, we decided not to compare experimental and binding scores in this study. Instead, we employed statistical analysis that combined RMSD, Z-score (Z(E)), and individual discriminative power (DPi) to evaluate the ability of the scoring functions to differentiate between well-docked and misdocked structures [21]. Discriminative ability is recognised as an important factor in assessing the accuracy of docking algorithms and scoring functions [33, 34]. The combination of the following equations of RMSD, Z-score, and DPi was used to calculate the Discriminative Power (DP) of a scoring function over the selected DNA-ligand complexes.
RMSD is the measurement of the average distance between the equivalent atoms of the docked binding mode and native crystallographic mode of the ligands.
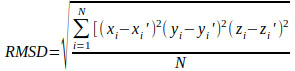 |
where (xi, yi, zi) and (xi′, y i′, z i′) are the Cartesian coordinates of the equivalent atoms in docked and native binding modes.
Z-score is the measurement of how many standard deviations are below or above the population mean of a given binding score:
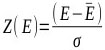 |
where E represents the binding score of the given DNA-ligand complex; Ē is the mean binding score over the set of docked ligand geometries; and σ is the standard deviation of the binding score distribution.
Individual Discriminative Power (DPi) of a given set of DNA-ligand complexes and Discriminative Power (DP) of a given scoring function over all of the selected DNA-ligand complexes is defined as:
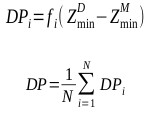 |
where fi is the fraction of the well-docked mode with Z-scores lower than that of the lowest Z-score misdocked mode, and Z Dmin and Z Mmin are the Z-scores of the lowest-energy well-docked and misdocked modes, respectively. Misdocked modes are those with an RMSD from the native mode of larger than 4Å, while well-docked modes are those with an RMSD from the native of less than 2Å. A DP of zero means that there is no discriminative power, while the lower the DP value, the more reliable the scoring function.
3. RESULTS AND DISCUSSION
3.1. Selection of DNA Complexes
In this study, 72 complexes were downloaded from the Protein Data Bank (PDB) using the keyword “DNA-ligand” in the search. Among them, 9 did not contain a ligand, 15 were generated using NMR methods, 2 had a resolution >2.5Å, 5 had an R-factor >0.25, 2 contained a ligand with a cofactor or substrate located within 5.00Å, and 6 of the ligands were solvent or buffer components. According to the mentioned filtering rules, these complexes were excluded. Hence, 33 out of the 72 complexes met all of the criteria for the docking evaluations in this study.
Ligands may bind to DNA bases in different types of modes; the most common are groove binding, intercalation binding and covalent binding. These bonds are composed of different types and strengths of intermolecular interactions, including hydrogen bonding, Van der Waals interactions, stacking interactions, etc. Scoring functions calculate the magnitude of these interactions using their own specific equations with different weighing methods. Hence, it is reasonable to believe that the accuracy of a scoring function varies according to the different types of binding mode, and hence varies between different complexes [10]. Here, the mode of 33 DNA-ligand complexes was classified into either minor groove, intercalation or others. The PDB codes of minor groove DNA-ligand complexes are 1d63, 1eel, 2b0k, 2b3e, 2dbe, 2f8w, 459d, 2gvr, 3oie, 3u05, 3u08, 4z4b, 109d and 360d. The PDB codes of intercalation DNA-ligand complexes are 1l1h, 2gb9, 3ce5, 3em2, 3eqw, 3eru, 3es0, 3et8, 3eui, 3eum, 3nz7, 3qsc, 3qsf and 3nyp. Those classified under others have PDB codes: 2hri, 3cdm, 3t5e, 3uyh and 4da3.
Helix-shaped double-stranded DNA forms two kinds of grooves: the major groove and the minor groove. In the minor groove, ligands bind in the small groove and the other parts of the molecules may be inserted between the base pairs of DNA. As the minor groove is narrower than the major groove, ligands are closer and hence more likely to interact with the DNA, so most ligands bind to the minor groove rather than the major groove [35]. The strength of this type of binding mainly depends on the combination of hydrogen bonding, Van der Waals interactions and electrostatic interactions [36]. Intercalation is another common binding mode of small and rigid aromatic ligands between base pairs of DNA, in which π-stacking and stabilising electrostatic interactions are the major mechanism of binding [37].
3.2. Binding Mode Prediction
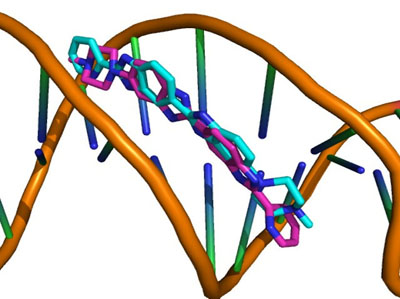
Each docking algorithm and its associated scoring functions generated fifty decoys for each of the 33 DNA-complexes. The average percentage of these 50 decoys with an RMSD less than 2.0Å (well-docked) and higher than 4.0Å (misdocked) in relation to their associated ligand crystallographic structures among the four scoring functions was 6.83% and 78.97%, respectively. This indicates that most of the decoys were misdocked structures. For the top 10 scored decoys, the average percentage with an RMSD <2.0Å and >4.0Å was 11.06% and 74.85%, respectively, with the four scoring functions performing better in the minor groove complex group than in the intercalated complex group, as indicated by 20.00% and 4.29% with an RMSD <2.0Å, respectively. These low percentage values indicate that the conformations of most of the top 10 favourable decoys were considerably different from the crystal structures. Among the four scoring functions, AutoDock obtained the highest number of well-docked decoys in the minor groove complex group, indicated by the percentage of 52.86% (Table 1), whereas ASP and ChemScore had the lowest performance (Table 2). For the intercalated complex group, the performance of the four scoring functions was low, ranging from 0.00% to 7.86% (Table 2). Examples of poor performance can be perceived on the 3qsc and 3qsf (Table 1). They are crystal structures of human telomeric DNA quadruplexes with salphen metals (NiII, CuII, PtII and VO2+) that are in line with potassium ions. These metals and ions form a complicated system in which π-π stacking energy, polarisation energy, and charge transfer are important for accurate modelling [38]. Generally, the development of specialised potential is required to model such systems; for example, the AutoDock4Zn is used to describe the interactions of zinc-coordinating ligands for zinc metalloprotein dockings [39]. Hence, the non-specific force field and docking approaches in this study could be the cause of the poor binding mode predictions in these two DNA complexes. Another example of poorly performing complexes include 3ce5, 3et8 and 3ny. These complexes contain ligands with an acridine functional group linked to two or three C-N bonds and C-C bonds, which can rotate and cause the docked confirmations to twist out of the planar surface of the native ligand, leading to the poor prediction of binding mode (Fig. 1).
| PDB code | Percentage of decoys with RMSD <2.0 Å | Percentage of decoys with RMSD >4.0 Å | ||||||
|---|---|---|---|---|---|---|---|---|
| ASP | ChemScore | GoldScore | Autodock | ASP | ChemScore | GoldScore | Autodock | |
| 1d63 | 0 | 10 | 40 | 70 | 90 | 70 | 30 | 30 |
| 1eel | 20 | 10 | 10 | 0 | 80 | 60 | 60 | 50 |
| 2b0k | 10 | 0 | 20 | 100 | 90 | 80 | 70 | 0 |
| 2b3e | 10 | 30 | 20 | 0 | 80 | 60 | 70 | 100 |
| 2dbe | 10 | 0 | 10 | 100 | 80 | 70 | 80 | 0 |
| 2f8w | 0 | 0 | 10 | 0 | 100 | 100 | 90 | 100 |
| 2gvr | 0 | 0 | 10 | 30 | 90 | 100 | 90 | 70 |
| 3oie | 0 | 0 | 10 | 70 | 90 | 100 | 70 | 30 |
| 3u05 | 20 | 10 | 10 | 100 | 60 | 60 | 60 | 0 |
| 3u08 | 10 | 10 | 20 | 100 | 80 | 80 | 70 | 0 |
| 4z4b | 0 | 0 | 0 | 0 | 100 | 100 | 100 | 100 |
| 109d | 20 | 10 | 20 | 100 | 80 | 80 | 70 | 0 |
| 360d | 0 | 10 | 10 | 70 | 100 | 90 | 60 | 0 |
| 459d | 0 | 0 | 10 | 0 | 100 | 100 | 60 | 100 |
| Mean | 6.43 | 6.43 | 14.29 | 52.86 | 87.14 | 82.14 | 70.00 | 41.43 |
| PDB code | Percentage of decoys with RMSD <2.0 Å | Percentage of decoys with RMSD >4.0 Å | ||||||
|---|---|---|---|---|---|---|---|---|
| ASP | ChemScore | GoldScore | Autodock | ASP | ChemScore | GoldScore | Autodock | |
| 1l1h | 10 | 0 | 20 | 50 | 40 | 100 | 20 | 40 |
| 2gb9 | 10 | 0 | 10 | 0 | 60 | 100 | 70 | 90 |
| 3ce5 | 0 | 0 | 0 | 0 | 30 | 80 | 50 | 100 |
| 3em2 | 0 | 0 | 10 | 30 | 50 | 100 | 40 | 50 |
| 3eqw | 0 | 0 | 10 | 0 | 100 | 100 | 30 | 50 |
| 3eru | 0 | 0 | 10 | 0 | 80 | 100 | 40 | 60 |
| 3es0 | 0 | 0 | 0 | 10 | 90 | 100 | 90 | 50 |
| 3et8 | 0 | 0 | 0 | 0 | 100 | 100 | 80 | 40 |
| 3eui | 0 | 0 | 10 | 0 | 70 | 100 | 60 | 90 |
| 3eum | 0 | 0 | 20 | 0 | 60 | 100 | 30 | 90 |
| 3nyp | 10 | 0 | 0 | 0 | 80 | 70 | 30 | 50 |
| 3nz7 | 0 | 0 | 20 | 10 | 50 | 100 | 20 | 70 |
| 3qsc | 0 | 0 | 0 | 0 | 100 | 100 | 100 | 100 |
| 3qsf | 0 | 0 | 0 | 0 | 100 | 100 | 100 | 100 |
| Mean | 2.14 | 0.00 | 7.86 | 7.14 | 72.14 | 96.43 | 54.29 | 70.00 |
The percentage of 1st ranked conformations with an RMSD less than <2.0Å over the 33 complexes among the four scoring was 17.42%. Again, AutoDock performed the best, with 50.00% for the minor groove and 21.43% for the intercalated complexes (Table 3). The percentage of 1st ranked conformations that were misdocked structures of ASP and ChemScore was the same: 92.86%. This shows that the ability of the docking and scoring methods to predict the binding mode of DNA-ligand complexes was low. Out of the 14 minor groove complexes, the performance of 4z4b was the worst. None of the four tested docking and scoring functions generated decoys with an RMSD <2.0Å (Table 1). Fig. (2) shows the experimental and AutoDock generated 1st ranked conformation structures of the DNA-complex 4z4b; it is clear that the misdocked structures are in the upside down position in the crystal structures. This misdocked structure is relatively stable, having a binding energy of -12.43 kcal/mol, which is close to the -12.00 kcal/mol of the crystallographic structure. The RMSD of all 50 decoys ranged from 11.38 to 11.40Å. This means that the ligands were trapped in a local minima under the AutoDock default parameter settings. Similar situations were also found in the other three docking and scoring programmes.
| DNA-ligand Complexes | Scoring Functions | |||
|---|---|---|---|---|
| ASP | ChemScore | GoldScore | Autodock | |
| RMSD <2.0 Å | ||||
| All | 9.09 | 9.09 | 18.18 | 33.33 |
| Minor groove | 21.43 | 14.29 | 28.57 | 50.00 |
| Intercalated | 0.00 | 7.14 | 14.29 | 21.43 |
| RMSD >4.0 Å | ||||
| All | 87.88 | 84.85 | 63.64 | 54.55 |
| Minor groove | 78.57 | 71.43 | 64.29 | 42.86 |
| Intercalated | 92.86 | 92.86 | 50.00 | 57.14 |
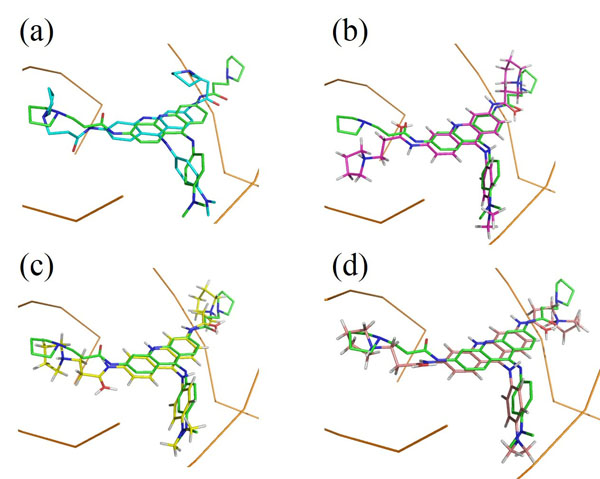
Another interesting result is the binding mode predictions of 2dbe, in which ASP, ChemScore and GoldScore obtained poor performances, while AutoDock obtained 100% of the top 10 ranked conformations with an RMSD <2.0Å (Table 1). Figs. (3a) and (3b) show the binding poses of the AutoDock and ASP 1st ranked confirmations, respectively. The speciality of this complex is its ligand-water interactions; the native crystallographic structure of the ligand shows that one of the amidinum groups of the ligand does not interact with the DNA directly; instead, a water molecule intervenes [40]. As for many docking procedures found in literature, all of the explicit water molecules in this study were deleted before docking. Hence, this explicit ligand-water-DNA interaction was missing in our results, leading to poor RMSD values. However, AutoDock somehow managed to model this water-mediated DNA-ligand interaction implicitly through the combination of its Lamarckian genetic docking algorithm and AutoDock4.2 force field [41]. Water has been proven to be important for some water-mediated protein-ligand interactions, and scoring functions incorporated with active-site water thermodynamic calculations, such as AutoDock-GIST [42], have been developed. This kind of function may also be useful in DNA-ligand complexes with many water molecules within the binding site.
The results of this study contrast with the study of Ricci et al. [43], who applied their docking protocols with DNA artificial conformational modifications to two DNA-ligand complexes using AutoDock. Their docked conformations were in strong agreement with the experimental binding mechanism, with 60-75% of the top ranked conformations obtaining an RMSD <2.00Å. In our study, only 11 of the 33 DNA-ligand complexes generated the top scored confirmation with an RMSD <2.0Å, while 5 DNA-ligand complexes had the top 10 scored conformations with an RMSD <2.0Å. The difference between the study of Ricci et al. and the current one may be due to the different sample sizes and docking approaches used. Ricci et al. performed dockings with their specific modifications on two DNA-ligand complexes, whereas this study involved 33 complexes and was performed using the default parameters. Hence, we believe that the protein-ligand parameterised AutoDock is capable of generating accurate docking results with specific modifications, and not with the default parameters. Through literature searches, the authors noticed that many studies performed DNA-ligand dockings using the programmes’ default parameters without performing any evaluation of their accuracy [44, 45]. This may lead to mistakes in predicting the binding mode.
3.3. Discriminative Power
Apart from predicting the binding mode, docking and scoring have also been used to select ‘true’ binders to aid in the drug discovery process. Hence, the ability of a scoring functions to distinguish between the native experimental confirmations among decoys is important; this discriminative power can serve as an evaluation method of accuracy [46]. Here, we evaluated the discriminative power of the four scoring functions over the compiled set of 200 (4 x 50) decoys for each of the 33 DNA-ligand complexes. ChemScore performed the best with a DP value of -0.27 and AutoDock had the lowest discriminative power of -0.14 (Table 4). However, it is interesting that ChemScore obtained a DP value of only -0.01 over the 50 decoys generated by itself (Table 5), and 30 out of the 33 complexes had a DPi value of zero, i.e. ChemScore had no discriminative power for 30 of them. The reason for such poor performance is that the fi value of most ChemScore DP calculations is zero. A zero fi value means there is no well-docked confirmation (RMSD <2.00Å), which obtained a Z-score lower than that of the lowest misdocked confirmation Z-score (RMSD >4.00Å). Hence, ChemScore tends to over-predict the binding affinities of the misdocked confirmations generated by itself. Vice versa, AutoDock obtained a DP value of -0.14 among the whole set of decoys, but performed much better in its own decoy set, indicated by a DP value of -0.46. This is because AutoDock tends to over-estimate the binding affinities of the decoys generated by other docking programmes.
| Discriminative Power (DP) | |||
|---|---|---|---|
| Scoring Functions | All Complex | Minor Groove | Intercalated |
| ASP | -0.22 | -0.17 | -0.22 |
| ChemScore | -0.27 | -0.27 | -0.32 |
| GoldScore | -0.16 | -0.25 | -0.13 |
| Autodock | -0.14 | -0.19 | -0.02 |
| Scoring Functions | Rescoring Functions | |||
|---|---|---|---|---|
| Autodock | ASP | ChemScore | GoldScore | |
| All decoys | ||||
| ASP | -0.01 | -0.01 | -0.11 | -0.20 |
| ChemScore | -0.07 | -0.33 | -0.01 | -0.21 |
| GoldScore | -0.02 | -0.28 | -0.28 | -0.03 |
| Autodock | -0.46 | -0.26 | -0.65 | -0.22 |
| Minor groove decoys | ||||
| ASP | -0.03 | -0.03 | -0.20 | -0.20 |
| ChemScore | -0.13 | -0.17 | -0.03 | -0.20 |
| GoldScore | -0.04 | -0.09 | -0.14 | -0.05 |
| Autodock | -0.55 | -0.37 | -0.73 | -0.41 |
| Intercalated decoys | ||||
| ASP | 0.00 | -0.13 | -0.05 | -0.10 |
| ChemScore | -0.01 | -0.50 | 0.00 | -0.30 |
| GoldScore | -0.01 | -0.25 | -0.53 | -0.01 |
| Autodock | -0.05 | -0.13 | -0.68 | -0.11 |
In addition to the DP analysis of each scoring function over the 200 decoys for each of the 33 DNA-ligand complexes, DP values were also calculated for the 50 decoys generated by each scoring function and were then rescored by another function. For example, the 50 decoys generated by ASP were rescored by AutoDock and the DP values were then calculated (Table 5). Among the scoring and rescoring, the ChemScore rescored AutoDock decoys obtained the best DP values of -0.65. This is significantly higher than that of the AutoDock and ChemScore with no rescoring, with results of -0.46 and -0.01, respectively (Table 5). This is due to the average enhancement of the individual DP values (DPi) over all DNA-ligand complexes. Among the 33 complexes, AutoDock, ChemScore and AutoDock rescored by ChemScore showed 21, 29 and 9 complexes with zero DPi values. Hence, rescoring AutoDock decoys with ChemScore improved the discriminative power of the 20 complexes from zero to acceptable. Among them, the DP values of complex 2b3e improved the most, changing from 0 to -4.18. 2b3e is a minor groove complex, where AutoDock calculated the binding affinity of the native crystallographic structure and the best misdocked conformation to be -11.79 and -12.04 kcal/mol, respectively. Hence the misdocked conformation is predicted to be more stable than the native structure. Vice versa, ChemScore rescoring predicted the native conformation to be more stable than the best misdocked binding mode, as indicated by the binding scores of 23.04 and 15.14, respectively. For AutoDock, more negative binding affinities indicate that the conformation is more stable. For ChemScore, high positive scores indicate stable conformations.

Another interesting result is the improvement of discriminative power when rescoring ChemScore decoys with ASP, indicated by the DP values of -0.01 and -0.33, respectively (Table 5). As when using ChemScore to rescore AutoDock decoys, ASP ranked the native conformation with a much higher score than the misdocked ChemScore decoys. ChemScore improved the DPi values of 11 complexes, with five of them showing DPi values which improved by at least 1.0: 1l1h, 3eru, 3eum, 3nz7 and 3uyh.
Many studies have demonstrated that rescoring can improve the accuracy of a scoring function on modelling protein-ligand interactions. These rescoring methods are generally more computationally intensive, such as MM/PBSA [47], QM/MM calculations [48], and specific binding free energy calculations [49]. Here, we demonstrated that the discriminative power of a scoring function on modelling DNA-ligand interactions can also be improved by rescoring (Table 5). This is probably due to the differences in approximate mathematical potentials to predict the strength of the intermolecular interactions, and their dissimilarity in parameterisation. For instance, AutoDock is an empirical-based free energy scoring function, which was parameterised using the inhibition constants (Ki) of a large number of protein-inhibitor complexes. The AutoDock scoring potential involves six pair-wise evaluations and an estimate of the conformational entropy lost upon binding. Each evaluation takes into account the pair-wise energetic terms including dispersion/repulsion, H-bonding, electrostatics, and desolvation [27]. The GoldScore fitness function was developed to predict protein-ligand binding modes through accounting factors, such as van der Waals energy, ligand torsion strain, H-bonding energy and metal interactions [28]. ChemScore fitness functions were parameterised based on the binding affinity data of 82 protein-ligand complexes. ChemScore integrates the total free energy change (ΔG) with a protein-ligand atom clash term and an internal energy term. The calculations of ChemScore also comprise H-bonding energy, metal interaction, ligand flexibility and hydrophobic-hydrophobic contact areas, etc [24]. The ASP fitness functions were augmented with the ChemScore clash term and internal energy term; this is an atom-atom distance potential derived from the protein-ligand database, as stated in the ASP reference [26]. The differences in the above scoring function somehow compensated for the deficiencies in individual scoring function while rescoring [22, 23].
Many studies have explored the discriminative powers of different scoring functions against different protein-ligand complexes. For example, Fong et al. [21] found that the DP values of ChemScore@GOLD and GoldScore@GOLD on six HIV-1 protease were -1.34 and -1.30, respectively. Brooks et al. [50] found the DP values of ChemScore@Sybyl and AutoDock on fifty-two aspartic proteases to be -1.45 and -0.88, respectively. Despite the different sample sizes, these DP values are superior to those found in the current study (Tables 4 and 5). This may indicate that docking and scoring approaches using the default parameters are more appropriate when modelling protein-ligand interactions than DNA-ligand interactions.
CONCLUSION
The literature suggested that protein-ligand docking accuracy can vary from 0% to 92.66% [10], and is highly dependent on the docking and scoring methods. Combining the results of this study and those in the literature, we believe that certain docking protocols and scoring functions, including AutoDock, ASP, ChemScore and GoldScore, are capable of reproducing the experimental crystallographic structure of DNA-ligand complexes using specific protocols [7, 9, 43]. However, most in silico approaches have not been parameterised for DNA-ligand interactions [51]; the commonly used docking approaches with default parameters may be suitable for protein-ligand complexes, but may not be appropriate for DNA-ligand complexes. Researchers should also consider the treatment of metals and water molecules in the active-site, especially those which have explicit ligand-water-DNA interactions. The difference between discriminative powers of scoring functions among the 33 DNA-ligand complexes in this study suggests that the evaluation of accuracies, such as binding mode identification, binding affinity prediction, and virtual database screening [15], should be performed for each DNA-ligand complex in order to obtain meaningful results in the drug discovery process.
In general, using the experimental crystallographic structures as the native structures to calculate RMSD is considered a useful indicator of docking accuracy in protein-ligand docking; however, this may not be the case for DNA-ligand complexes as they are very flexible and may have several isoforms [52, 53]. Hence, the binding modes of the downloaded crystallographic structures may not be the only ‘real’ binding mechanisms. Comparing the docked structures with these crystallographic structures may lower the true accuracy of the docking algorithms and scoring functions. This could be a limitation of the current study.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
No Animals/Humans were used for studies that are the basis of this research.
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
FUNDING
This work was supported by the Macao Polytechnic Institute Research Fund (Project No: RP/ESS-04/2016).
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
Declared none.

