All published articles of this journal are available on ScienceDirect.

The Utility of Oligopeptidase in Brain-Targeting Delivery of an Enkephalin Analogue by Prodrug Design
Abstract
In a brain-targeting prodrug approach for a metabolically stable enkephalin analogue DADLE, specific enzymes are utilized for in vivo prodrug activation. Prolyl oligopeptidase (POP) may be especially useful in this regard. In vitro metabolic stability of the putative metabolites of prodrugs having various “spacers” has shown that POP provides significantly faster release of DADLE from conjugates having dipeptidyl spacer (specifically Xaa-Pro or Xaa-Ala) than alternative peptidases utilized when single amino acids are used as spacers. In vitro half-lives measured in rat brain homogenate showed excellent correlation with CNS-mediated analgesia using the tail-flick model in rats providing, thus, an in vivo substantiation of the prodrug approach relying on POP as the peptidase to release DADLE.
INTRODUCTION
Enkephalins are opioids and potential neuropharmaceuticals for pain relief without the undesired side-effects associated with the use of morphine and other alkaloid analgesics [1]. These endogenous linear pentapeptides are, however, easily degraded by peptidolytic enzymes [2] rendering their action short-lasting, unless peptidase inhibitors are used simultaneously [3]. Therefore, a wide variety of analogues with increased in vivo stability have been designed and evaluated. A (δopioid receptor-selective enkephalin analogue Tyr-D-Ala-Gly-Phe-D-Leu (DADLE), is a representative of such compounds [4]. In spite of adequate metabolic stability of DADLE and the majority of synthetic neuropeptide analogues, these agents still face a formidable obstacle, the blood brain barrier (BBB), in terms of reaching the central nervous system (CNS) upon systemic administration [5]. The BBB, acting as a lipoidal bilayer, practically excludes the entry of these hydrophilic substances into the brain by passive transport. Additionally, BBB may also represent an enzymatic barrier for peptides. As such, the limited bioavailability of the otherwise promising peptide-based neuropharmaceuticals presents a major disadvantage for practical drug development.
Invasive and non-invasive strategies have been developed to outwit the BBB [6]. Among non-invasive methods, prodrug design is perhaps the most versatile chemical manipulation technique that bioreversibly modifies the parent peptide for improving CNS bioavailability and targeting. A specific approach within prodrug design is the use of a brain-targeting chemical delivery system (BTCDS) [7]. BTCDS, as a prodrug, is biologically inactive per se [8] and requires in vivo enzymatic conversion(s) to the biologically active agent and provides enhanced drug delivery to the brain, the site of action. In peptide-based BTCDS, the lipophilic 1,4-dihydrotrigonellyl transport moiety [7] is covalently attached to the N-terminus of the parent peptide via “spacer” amino acid(s). In the brain, however, a coenzyme associated with numerous oxidoreductases and cellular respiration oxidizes the dihydropyridine to permanently charged N-methylpyridi-nium (trigonellyl, T+), analogously to the oxidation of NAD (P)H to NADP+ [7]. The “oxidized” BTCDS apparently cannot leave the brain by passive transport, and it has not been identified as a potential substrate for endogenous efflux transporters either [9]. A strategically selected spacer ensures, then, the release of the parent peptide in the brain by specific peptidases [10, 11] that remove the T+-Spacer moiety [11] from the “oxidized” BTCDS.
Previously, we have reported a successful application of this approach to transport DADLE [11, 12] into the brain (Fig. 1). The C-terminus of DADLE was esterified with highly lipophilic alcohols, such as cholesterol (Ch-OH) to furnish adequate lipophilicity of the BTCDS necessary to BBB transport [9]. This ester group can easily be cleaved by lipases and/or esterases during or after diffusion through the BBB. Alanine (Ala) or proline (Pro)-based spacer (S) was originally chosen because of the suggested involvement of alanyl and prolyl aminodipeptidyl peptidases (EC 3.4.14.2 and/or EC 3.4.14.5) [13] in the enkephalinergic transmission in the brain. However, as it turned out [14], these enzymes can also be utilized for the release of other neuropeptides, such as thyrotropin releasing hormone, from the corresponding “oxidized” BTCDS. Additionally, peptidyl glycine (-amidating monooxygenase (PAM) may also be exploited for peptide-based brain-targeting prodrug design [14,15].
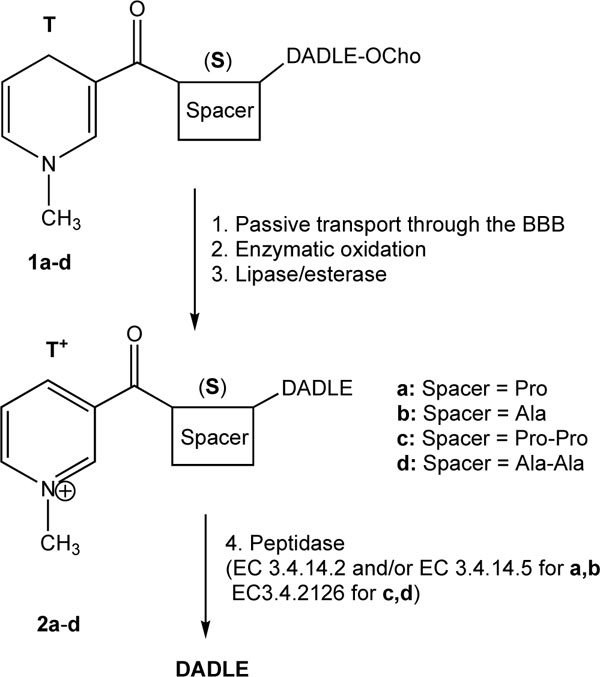
Schematic illustration of a BTCDS for DADLE, Tyr-DAla-Gly-Phe-D-Leu. OCho represents cholesteryl ester [12].
In vivo substantiation of the approach for DADLE shown in Fig. (1) was done by monitoring the antinociceptive effect produced after intravenous (iv) administration of BTCDSs in rats. Although BTCDSs containing either Pro or Ala spacer (1a and 1b, respectively) showed a profound, centrally-mediated and long-lasting analgesia, 1a was more potent than 1b in terms of producing a quicker onset and longer duration of tail-flick latencies [12]. Consequently, we concluded that the “spacer” was the most important architectural element of the BTCDS (1a-d) to manipulate the rate of DADLE release and, thus, the production of analgesia by utilizing specific peptidases (probably EC 3.4.14.2 and/or EC 3.4.14.5). We have also reasoned that another type of enzymes such as prolyl oligopeptidase (POP, EC 3.4.21.26, that has specific cleaving sites in a peptide sequence [16] may also be used to control peptide release from the oxidized BTCDSs according to the therapeutic objectives.
The present studies were undertaken to provide in vitro evidence of this hypothesis [17], together with an in vivo investigation of the pharmacological consequences of introducing Pro-Pro (1c) and Ala-Ala (1d) dipeptidyl spacers into BTCDSs for potential exploitation of POP [16] in brain-targeting delivery of DADLE by prodrug design.
SYNTHESIS
Selected BTCDSs having Pro-Pro or Ala-Ala spacer and Cho ester on their C-termini (1c and 1d, Fig. 1) were synthesized analogously to that of 1a and 1b (S=Pro and S=Ala, respectively)by a segment coupling method we have developed to obtain these types of peptide conjugates [12]. Briefly, Nicotinoyl-(S)-D-Ala-Gly-Phe-OH was prepared by routine Fmoc-based solid phase peptide synthesis (SPPS) in a Synthor 2000 instrument (Peptide International, Louisville, KY) and then coupled to D-Leu-OCho in solution phase using PyBOP/HOBt/DIPEA activation The latter lipophilic ester of the C-terminal amino acid (D-Leu-OH) was prepared by solution-phase Boc-chemistry. Once Nicotinoyl-(S)-DADLE-OCho was assembled, column chromatographic purification (silicagel, chloroform/methanol 9:1, v/v) was done. Then the nicotinoyl residue was converted to trigonellyl (T+) using MeI. After the well-established reduction using Na2S2O4 [18] the BTCDSs were obtained. This reductionwas conveniently monitored by UV spectroscopy (Lambda 11 UV/Vis Perkin Elmer spectrometer; Perkin Elmer Anal. Inst., Norwalk, CT) in methanol that showed the unequivocal conversion of T+ (λ=254 nm) to 1,4-dihydropyridine (T) (λ=355 nm). The putative oxidized metabolites 2a-e from which DADLE may be directly released were also synthesized by routine Fmoc-based SPPS. As control, 2g having no spacer moiety was also prepared. All test compounds were thoroughly purified by reversed-phased high performance liquid chromatography (RP-HPLC) and identified by electrospray-ionization mass spectrometry (ESI-MS), as described before [12].
IN VITRO STABILITY STUDIES
Stability studies of the predicted biotransformation products, T+-S-DADLE, of the BTCDSs have been performed in rat brain homogenate [17]. Approximately 250 nmol of 2a-g, dissolved in 100 mL DMSO, was added to 1 mL of freshly made rat brain homogenate (20% w/w, pH 7.4 Tris buffer [19]). For comparison, T+-DADLE (2g), in which DADLE is directly attached to T+ was also studied to underline the importance of the “spacer” in the molecular architecture of such type of BTCDSs. The mixture was incubated at 37 °C in a temperature controlled, shaking water bath. Aliquots (100 µL) were taken after 5, 15, 30, 45, 60, and 90 min of incuba-tion, and transferred to 1.5 mL plastic centrifuge tubes containing 200 µL of ice-cold 1 M aqueous acetic acid. After centrifugation at 12500g for 15 min, the supernatant was removed and analyzed by microbore HPLC on a system consisting of a ThermoSeparation/Spectra-Physics (Fremont, CA) Spectra Series P200 binary gradient solvent delivery system, a Rheodyne (Cotati, CA) Model 7125 injector valve equipped with a 5 µL sample loop, a Spectroflow 757 variable wavelength UV/Vis detector (Kratos Analytical, Manchester, U.K.) operated at 216 nm.
As shown in Table 1, the conjugate without a spacer (2g) is extremely stable (t1/2 is almost 8 h); thus, no release of DADLE in the brain could be expected from the corresponding prodrug. This finding supports the notion that amidase activity is low in the brain [7]. When a single amino-acid spacer was used and prolyl and alanyl aminodipeptidyl peptidases were the apparent processing enzymes responsible for DADLE release, 2a (S=Pro) was approximately 3-times less stable than 2b (S=Ala). This observation is in agreement with our previous in vivo findings [12]; a more profound and earlier onset of analgesia was produced with Pro (1a) compared to that of Ala (1b) as a spacer (see Table 3).
Metabolic Stability of T+-S-DADLE and T+-DADLE in Rat Brain Homogenate
| Putative Metabolite | t1/2 (min) |
| T+-Pro-DADLE 2a) | 346 |
| T+-Ala-DADLE 2b) | 963 |
| T+-Pro-Pro-DADLE (2c) | 9 |
| T+-Ala-Ala-DADLE (2d) | 23 |
| T+-Pro-Ala-DADLE(2e) | 24 |
| T+-Ala-Pro-DADLE(2f) | 24 |
| T+-DADLE(2g) | 4620 |
Concentration–time profiles were analyzed by exponential fitting assuming pseudofirst order degradation. In vitro half-lives (t1/2) were calculated from rate constant (k) as 0.693/k.
Antinociception in Sprague-Dawley Rats After i.v.Injection (4.2 µmol/kg Body Weight Dose) of Prodrugs 1a-d of DADLE
| Prodrug | ~TMAX (min) | Anti-Nociceptiona |
| 1a S=Pro |
30 | 2.7 |
| 1b S=Ala |
60 | 2.1 |
| 1c S=Pro-Pro |
5 | 1.6 |
| 1d S=Ala-Ala |
5-10 | 1.8 |
aAntinociception was defined as (Maximum latency –Baseline latency)/Baseline latency.TMAX was defined as time (min) to reach maximum tail-flick latency.
When dipeptidyl spacers (specifically Xaa-Pro or Xaa-Ala) were used where POP is expected to be the processing enzyme [16], then the half-life of 2c (S =Pro-Pro) was especially short (<10 min), suggesting the very quick release of DADLE in the brain. Interestingly, in case of Ala-Ala (2d), Pro-Ala (2e) or Ala-Pro (2f) combinations, the in vitro half-lives were practically the same (around 20 min) that may imply that the affinity of POP to Pro is somewhat higher than to Ala, especially when Pro is the preceding amino acid. (Under our experimental conditions, t1/2 for DADLE was around 730 min). Nevertheless, these in vivo data clearly show that the metabolic stability and, thus, DADLE–releasing rate of the putative metabolites (2a-f) of the corre-sponding BTCDSs can be manipulated by strategically se-lecting the spacer function.
The in vitro release of DADLE and the concomitant loss of the T+-S moiety were unequivocally shown by ESI-MS analysis (data not shown) [12,17]. Altogether, it can be concluded that POP, indeed, can be utilized for the release of DADLE from the corresponding oxidized BTCDSs. The measured very short in vitro half-lives in brain homogenate may also predict a quick onset of analgesia upon i.v. admini-stration of the BTCDSs having dipeptidyl spacers.
The distribution of POP activity within the brain was also investigated by incubating 100 µM 2c (S=Pro-Pro) in homogenates of different brain sections (Table 2) analogously to the metabolic studies involving whole brain homogenate (Table 1). Among the studied brain sections, the highest metabolism was measured in the cortex, while 2c was quite stable (t1/2 > 1 h) in the medulla oblongata indicating low POP activity in this region of the brain. These observations are in agreement with findings about the uneven distribution of POP in the brain [20, 21]. Therefore, because POP is exclusively expressed by neurons and display region-specific distribution in the mouse brain, POP-activated prodrugs may be especially useful to target neurons in select regions (cortex, cerebellum, striatum and mid-brain) of the brain.
Metabolic Stability of T+-Pro-Pro-DADLE (2c) in Homogenates of Rat Brain Sections
| Brain Section | t1/2(min) |
| Cortex | 4 |
| Medulla Oblongata | 72 |
| Cerebellum | 9 |
| Striatum | 12 |
| Hypothalamus | 28 |
| Mid-brain | 12 |
| Whole brain | 9 |
Concentration-time profiles were analyzed by exponential fitting assuming pseudo-first order degradation.In vitro half-lives (t1/2) were calculated from rate constant (k) as 0.693/k.
ANALGESIC ACTIVITY
In vivo substantiation of the in vitro metabolic stability studies (Table 1) and, thus, DADLE release in the brain from 1c and 1d as representative prodrugs having dipeptidyl spacer was done by monitoring CNS-mediated analgesia in rats upon i.v. injection of the BTCDSs (in 1:9 v/v DMSO : 20% w/v aqueous hydroxypropyl-β-cyclodextrin) using the tail-flick model [11, 12, 22]. The latency of the tail-flick response to radiant heat (focused in the middle of the tail) was measured by using a Tail-Flick Analgesia Meter 0570-001L (Columbus Instruments, Columbus, OH, USA). Base-line latencies (5-6 s) were collected for 15 min prior to prodrug administration, and tail-flick latency was measured at various times up to 6 h following BTCDS administration. Groups of 5-10 animals were treated with vehicle control or the prodrugs at 4.2 µmol/kg of body weight dose. Data generated from the studies comparing treatment groups were subjected to analysis of variance (ANOVA) followed by Dunnett’s post hoc procedure to detect significant differences (P >0.05) between groups [23].
Administration of the dipeptidyl spacer-containing prodrugs (1c and 1d) produced analgesia significantly faster than prodrugs having single amino acid spacer (1a and 1b, respectively) as shown in Table 3. These data show excellent correlations with the measured in vitro half-lives (Table 1). The corresponding putative metabolites 2c and 2d were ap-proximately 40-fold less stable in vitro than 2a and 2b,respectively, although more potent antinociception was observed with the use of single amino acid spacers where dipeptidyl aminopeptidase was the processing enzyme.
In summary, our data clearly show that utilizing POP in brain-targeting prodrug design to transport peptides into the brain could be especially useful when the therapeutic objective calls for a very quick onset of action, thus, release of the parent peptide. We have also envisioned that, by optimizing the amino acid that precedes Pro or Ala in the dipeptidyl spacers shown here, the release of the biologically active peptide could further be controlled. These studies will be published separately.
ACKNOWLEDGEMENT
The authors conducted the studies presented here while working at the Center for Drug Discovery, University of Florida. Technical assistance of Zoltan Szeiler with the tailflick latency measurements and support by Nicholas Bodor during these studies are acknowledged.

