All published articles of this journal are available on ScienceDirect.

ROCK2 Inhibition Underlying the Anticancer Effects of Dobutamine: A Novel Proposed Mechanism
Abstract
Introduction
Dobutamine, a well-established β1-adrenergic agonist, has demonstrated anti-proliferative effects against several cancer cell types. However, the molecular mechanism underlying this activity has remained elusive. Collective evidence from the literature indicates that dobutamine-sensitive cancers-including osteosarcoma, gastric adenocarcinoma, and multiple myeloma-frequently overexpress Rho-associated protein kinase 2 (ROCK2). To evaluate the hypothesis that dobutamine can act as a ROCK2 inhibitor.
Methods
A comprehensive approach was employed, including enzymatic assays, cell-based proliferation studies in cell lines with differential ROCK2 expression, and computational molecular docking analyses.
Results
Enzyme assays demonstrated that dobutamine inhibits ROCK2 with a half-maximal inhibitory concentration (IC50) of 7.1 µM. In cellular assays, dobutamine induced a ROCK2 expression-dependent anti-proliferative effect, showing approximately fourfold greater potency against the high-ROCK2-expressing HepG2 cell line compared to the low-ROCK2-expressing T-47D cell line. Furthermore, molecular docking studies revealed a plausible ATP-competitive binding mode of dobutamine within the ROCK2 kinase domain, stabilized by key hydrogen bonding, hydrophobic interactions, and π-cation interactions.
Discussion
Our findings provide the first direct evidence that dobutamine inhibits ROCK2 enzymatic activity, as demonstrated by both enzyme-based and cell-based assays. Moreover, our results validate dobutamine's chemical scaffold as a promising starting point for the structure-based design of more potent and selective ROCK2-targeted anticancer agents. Notably, we identified two major thermodynamic limitations contributing to its suboptimal binding affinity: a high entropic penalty and an enthalpic penalty associated with desolvation. Accordingly, clear medicinal chemistry optimization strategies can be employed to overcome these issues.
Conclusion
This study establishes a novel mechanistic link between dobutamine and ROCK2 inhibition, providing a strong rationale for its therapeutic repurposing.
1. INTRODUCTION
The traditional de novo drug discovery and development pipeline is a challenging process, characterized by extensive timelines, prohibitive costs, and a high rate of attrition. In response to these challenges, drug repurposing-also known as drug repositioning or reprofiling-has emerged as a powerful and efficient strategy for therapeutic innovation. This approach identifies new therapeutic indications for existing drugs, capitalizing on established data regarding their safety, pharmacokinetics, and manufacturing processes to substantially shorten the bench-to-bedside trajectory [1]. The field of oncology, in particular, has been a fertile ground for drug repurposing, with numerous agents from diverse pharmacological classes being successfully repositioned to combat various malignancies [2].
Computational approaches, particularly molecular docking and machine learning-based methods, have become indispensable tools for drug repurposing, enabling rapid identification of novel drug-target interactions [3, 4]. The integration of artificial intelligence with structure-based virtual screening has proven especially valuable for kinase inhibitor repositioning, given the conserved architecture of ATP-binding pockets across the kinome [5]. The present study employs validated molecular docking protocols to complement our experimental investigations of the dobutamine-ROCK2 interaction.
Dobutamine (Fig. 1) is a synthetic catecholamine primarily known for its clinical use as a β1-adrenergic receptor agonist in the management of acute heart failure and cardiogenic shock [6]. Beyond its cardiovascular applications, a growing body of evidence has pointed towards unexpected anticancer properties. Seminal studies have reported that dobutamine can significantly suppress the growth of human osteosarcoma cells by inhibiting proliferation and inducing apoptosis [7]. Similarly, it has been shown to exert inhibitory effects on human gastric adenocarcinoma, impacting cell growth, colony formation, and invasion [8]. Furthermore, dobutamine has demonstrated antitumor activity against multiple myeloma cells, mediated by apoptosis induction, cell cycle arrest, and inhibition of the MAPK signaling pathway [9]. Despite these compelling phenotypic observations, the precise molecular target responsible for these anticancer effects has remained unidentified.
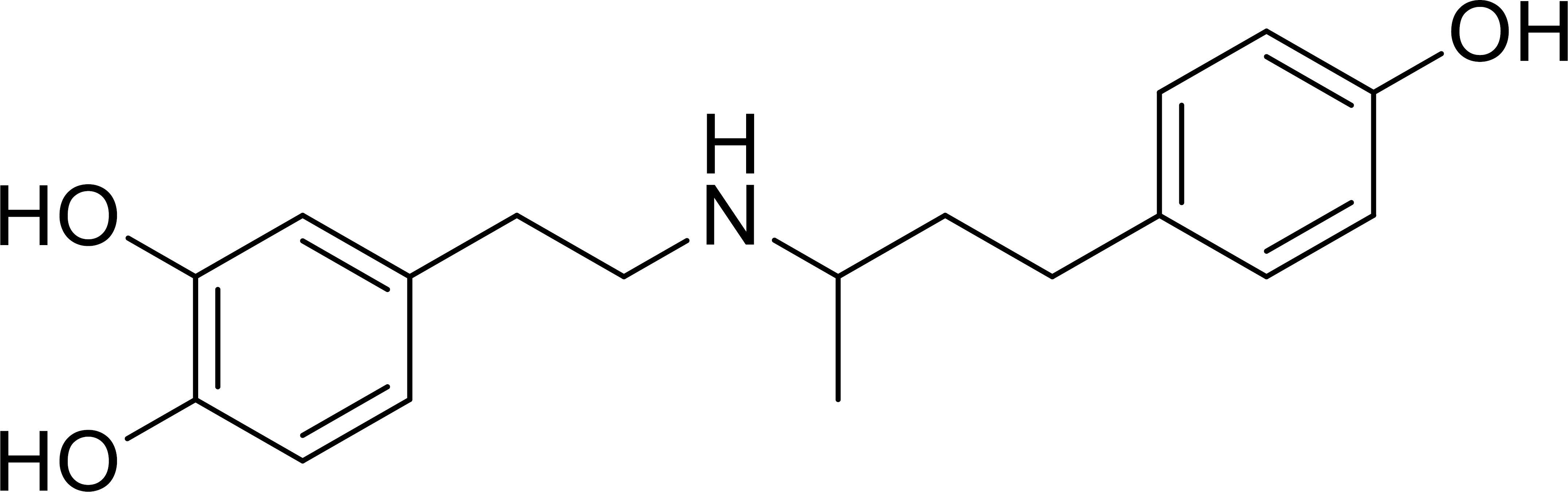
Chemical structure of dobutamine.
Rho-associated protein kinase 2 (ROCK2) is a serine/ threonine kinase that functions as a critical downstream effector of the RhoA GTPase. It is activated by GTP-bound RhoA, a process that is further enhanced by Polo-like kinase 1 (PLK1), which phosphorylates ROCK2 at additional sites to potentiate its activity [10, 11]. ROCK2 plays a pivotal role in regulating fundamental cellular processes, and its dysregulation is deeply implicated in cancer progression, promoting tumor proliferation, invasion, metastasis, and angiogenesis [12, 13]. A careful examination of the literature reveals that ROCK2 is not merely overexpressed but acts as an active driver of aggressive biology across the very cancer types sensitive to dobutamine, positioning it as a compelling “convergent hub” of malignancy [14, 15].
The therapeutic importance of targeting ROCK2 in oncology has gained substantial momentum in recent years. A comprehensive review by Ning et al. (2024) has elucidated the complex pathophysiological functions of RhoA-ROCK2 signaling in cancer progression, highlighting its role in epithelial-mesenchymal transition, migration, invasion, and therapy resistance associated with cancer stem cells [16]. Furthermore, the clinical translation of ROCK inhibitors has advanced considerably, with a recent patent review by Rodrigues de Oliveira et al. (2025) cataloging significant developments in ROCK inhibitor design from 2017 to 2023, underscoring the sustained pharmaceutical interest in this target class [17]. The field of kinase-targeted cancer therapy has also witnessed remarkable growth, with over 120 small-molecule kinase inhibitors now approved globally for treating various diseases, and nearly 70 FDA-approved specifically for cancer treatment as of 2024 [18]. This expanding therapeutic landscape provides a fertile context for the identification of novel ROCK2 inhibitors through drug repurposing strategies.
In osteosarcoma, ROCK2 is a key negative prognostic factor and a mediator of therapeutic resistance. Recent studies have demonstrated that elevated ROCK2 expression is associated with poor patient outcomes and drives resistance to treatments like photodynamic therapy by modulating autophagy [14, 15]. In gastric cancer, ROCK2 upregulation is correlated with increased tumor grade, invasion, and poor prognosis [19]. More recent mechanistic work has shown that ROCK2 stability is tightly regulated under hypoxic conditions and that its dysregulation is a key step in promoting epithelial-mesenchymal transition and metastasis [20]. In multiple myeloma, the therapeutic relevance of ROCK2 is well-established, with the selective ROCK2 inhibitor belumosudil demonstrating direct antimyeloma effects and enhancing the efficacy of other therapies [21]. This context is particularly relevant in the rapidly evolving treatment landscape for multiple myeloma, which now includes advanced quadruplet therapies and immunotherapies.
Recent advances have further validated ROCK2 as a promising anticancer target. Notably, Meloni et al. (2024) demonstrated that belumosudil exerts direct antimyeloma effects and enhances isatuximab-mediated cytotoxicity against multiple myeloma cells, providing compelling evidence for the clinical utility of ROCK2 inhibition in hematological malignancies [21]. Concurrently, preclinical studies by Pajic et al. (2024) have shown that combining zelasudil, a novel selective ROCK2 inhibitor, with chemotherapy or immunotherapy significantly improves response rates in pancreatic cancer models, further expanding the potential oncologic applications of ROCK2 inhibition [21, 22].
The convergence of dobutamine's unexplained anticancer activity in specific malignancies and the established role of ROCK2 as a critical oncogenic driver in those same cancers led us to formulate a central hypothesis: Dobutamine exerts its anticancer effects, at least in part, through the direct inhibition of ROCK2. To rigorously test this hypothesis, we designed a multi-pronged investigation comprising: (i) direct enzymatic assays to quantify the inhibitory effect of dobutamine on ROCK2 kinase activity; (ii) cell-based proliferation assays using cancer cell lines with differential ROCK2 expression to link target engagement with a cellular phenotype; and (iii) in silico molecular docking to elucidate the structural basis of the drug-target interaction and provide a blueprint for future optimization [1, 23-25].
2. MATERIALS AND METHODS
2.1. Enzymatic Inhibition Assay
The inhibitory activities of dobutamine against ROCK2 and PLK1 were evaluated using the Z'-LYTE Kinase biochemical assay (Invitrogen, USA). Dobutamine HCl was gifted by Hikma Pharmaceuticals, Amman, Jordan (batch number: 21100697). This assay employs a fluorescence-based, coupled-enzyme format and is based on the differential sensitivity of phosphorylated and non-phosphorylated peptides to proteolytic cleavage. The peptide substrate is labeled with two fluorophores, the donor (coumarin) and the acceptor (fluorescein), which form a FRET (fluorescence resonance energy transfer) pair. When the peptide is phosphorylated, it resists proteolytic cleavage, maintaining the FRET signal. However, if the kinase is inhibited and the peptide is not phosphorylated, it becomes susceptible to cleavage, resulting in the loss of the FRET signal. This enables sensitive and reliable detection of kinase inhibition [26].
Initially, the bioactivity of dobutamine against ROCK2 and PLK1 was assessed at a single concentration of 10 µM. If the inhibitory activity was promising (i.e., % inhibition at 10 µM > 50%), additional concentrations were tested to determine the IC50 value. Each concentration was tested in duplicate. All statistical analyses were performed using GraphPad Prism (Version 9.3.1, GraphPad Software, Inc., San Diego, CA). The IC50 value was calculated using nonlinear regression of log (concentration) versus percent inhibition. Staurosporine was used as a positive control, with IC50 values of 2.49 nM for ROCK2 and 617.0 nM for PLK1 [26].
The Z'-LYTE platform was selected for this study based on its established reputation as an industry-standard kinase profiling technology with demonstrated reliability across more than 200 kinases [27]. This FRET-based assay offers several advantages for kinase inhibitor characterization, including ratiometric quantification that minimizes compound interference, high reproducibility, and standardized conditions that facilitate cross-study comparisons [28]. The assay has been extensively validated in drug discovery applications for both lead identification and selectivity profiling, with the SelectScreen® service being widely utilized in pharmaceutical research [29]. Internal validation was achieved using staurosporine as a positive control, which yielded IC50 values consistent with published literature, thereby confirming assay performance under our experimental conditions.
2.2. Cell Proliferation Assay
Hep-G2 cells (ATCC® HB-8065™) and T-47D cells (ATCC® HTB-133™), were cultured in high-glucose Dulbecco’s Modified Eagle Medium (DMEM) fortified with 10% fetal bovine serum (FBS), sodium pyruvate (1 mM), 1% (v/v) L-glutamine (2 mM), 1% (v/v) penicillin (100 IU/mL) and streptomycin (100 μg/mL). Cells were seeded in 96-well plates at a density of 5.5*103 cells per well and incubated in a CO2 incubator at 37 ◦C. After 24 hours of seeding, dobutamine was added in a serial dilution in tissue culture media to achieve the concentrations of 250, 125, 62.5, 31.25, 15.625, 7.8125, 3.9, 1.95, and 0.485 µM. An incubation period of 72 hours followed, after which the MTT assay was carried out to assess cell viability and determine the IC50 values of the treatment. MTT dye was reconstituted using PBS at a 5:1 (w/v) ratio. After decanting the drugs and media, 100 μL of fresh medium in addition to 10 μL of the MTT mixture were added to each well, then the plates were incubated for 3-4 hours. The cells were examined under the microscope to ensure the formation of purple formazan crystals. Subsequently, the dye and medium were aspirated, and 100 μL/well of DMSO was added to solubilize the crystals. The plates were placed on the plate shaker for 15-20 minutes, and the absorbance was read at a wavelength of 560 nm using a microplate reader (Micro Read 1000, Global Diagnostics B, Belgium). Cell viability was calculated by comparing the absorbance of drug-treated wells to those of controls of no treatment. Each concentration was measured in quadruplicate wells, and the whole plate was repeated using at least two independent cell passages.
The MTT assay was employed as a well-established colorimetric method for assessing cell viability and proliferation, recognized for its extensive utility in cytotoxicity screening and drug discovery applications [30]. While the assay measures metabolic activity as a proxy for cell viability, careful optimization of assay parameters (cell seeding density, incubation time, and MTT concentration) was performed to ensure reliable quantification [31]. The selection of HepG2 and T-47D cell lines was based on their differential ROCK2 expression profiles as documented in the Human Protein Atlas transcriptomics database [32], providing an ideal experimental model to test the hypothesis that ROCK2 expression levels correlate with dobutamine sensitivity. This approach of comparing drug effects in cell lines with high versus low target expression is a recognized strategy for phenotypic validation of target engagement [33]. Experimental reliability was ensured through quadruplicate measurements, biological replication using independent cell passages, and appropriate controls for normalization.
2.3. Molecular Docking
Searching the protein databank (https://www. rcsb.org/) identified 17 crystallographic structures of Homo sapiens ROCK2, of which only two had reasonable resolution (i.e., less than 2.5 Å). The crystal structure with the lowest resolution (PDB code: 8X92, resolution 2.2 Å) was selected for the docking experiment.
The selected crystal structure was downloaded and prepared for docking using the protocol “Prepare protein” implemented in Discovery Studio (Version 4.5, Biovia Inc., USA). The following amino acids were completed and energy refined: LYS29, GLU31, ARG35, ARG38, LYS64, LYS66, ASN70, GLU76, LYS77, LYS80, LYS81, ARG100, LYS130, LYS189, LYS310, GLU374, GLU388, ASP389, and LYS401. Hydrogen atoms were added to the proteins utilizing Discovery Studio templates for protein residues. The resulting protein structures were used in subsequent docking experiments without further energy minimization. LibDock was used as a docking engine, and the binding site was defined based on the co-crystallized bound ligands, YEW.
The site-feature docking algorithm, LibDock, inserts ligands, after removing hydrogen, into a predicted active site based on binding hotspots. It aligns ligand conformations by matching their functional groups with polar and nonpolar interaction sites (hotspots) within the receptor. Ligand conformations can be generated during the docking process or pre-generated. A CHARMm minimization step can be optionally applied to refine the docked poses [34, 35].
Initially, self-docking was performed, where the co-crystallized ligand was docked into the ROCK2 binding site. Subsequently, all resulting docked poses were scored using seven scoring functions: JAIN [36], LigScore1, LigScore2 [37], PLP1 and PLP2 [38], PMF, and PMF04 [39]. The top-scoring docked poses from each of these functions were compared to the co-crystallized pose to identify the best one that could reproduce the crystal-bound pose. Self-docking was performed to select and validate the docking settings, and subsequently determine the most probable pose of dobutamine within the ROCK2 binding site.
The following LibDock parameters were used in this study: the binding site radius was set to 10.2 Å, and the docking tolerance to 0.25 Å. A total of 100 hotspots were defined. The conformation generation option was set to CAESAR, generating up to 250 conformers that did not exceed an energy threshold of 20 kcal/mol above the most stable conformer. The docking preference was set to “High Quality”.
The molecular docking protocol was validated through self-docking (cognate docking) of the co-crystallized ligand YEW, a standard procedure for verifying that docking parameters and scoring functions can accurately reproduce experimentally determined binding poses [40]. The protocol was considered validated when the root mean square deviation (RMSD) between the docked pose and the crystallographic pose was below the widely accepted threshold of 2.0 Å [41, 42]. The use of multiple consensus scoring functions (JAIN, LigScore1, LigScore2, PLP1, PLP2, PMF, and PMF04) is recognized as a best practice to enhance the reliability of pose prediction and reduce scoring function bias [43]. This consensus approach has been shown to improve virtual screening performance by mitigating the limitations inherent to individual scoring functions [44].
2.4. Statistical Analysis
All statistical analyses were performed using GraphPad Prism (Version 9.3.1, GraphPad Software, Inc., San Diego, CA). For enzymatic inhibition assays, IC50 values were calculated by nonlinear regression fitting of log (concentration) versus percent inhibition using a four-parameter logistic equation; each concentration was tested in duplicate. For cell proliferation assays, each drug concentration was tested in quadruplicate wells (technical replicates), and experiments were independently repeated using at least two different cell passages (biological replicates). IC50 values are reported with 95% confidence intervals (CI). Statistical significance between cell lines was assessed by the non-overlapping confidence interval method [45], with p < 0.05 considered statistically significant. Data are presented as mean ± standard deviation (SD) where applicable.
3. RESULTS AND DISCUSSION
The conclusions of this study are supported by the convergence of three independent lines of evidence. First, biochemical assays demonstrated direct, dose-dependent inhibition of recombinant ROCK2 (IC50 = 7.1 µM) with a Hill slope of 0.85, consistent with well-behaved single-site inhibition. Second, cellular assays revealed a statistically significant correlation between ROCK2 expression levels and sensitivity to dobutamine's antiproliferative effects, with high-ROCK2-expressing HepG2 cells showing 3.7-fold greater sensitivity than low-ROCK2-expressing T-47D cells (p < 0.05). Third, molecular docking studies, validated by successful reproduction of a known co-crystal structure (RMSD = 1.27 Å), identified a plausible ATP-competitive binding mode featuring key hydrogen bonds with hinge-region residues and engagement of the catalytic lysine. Together, these data establish a novel mechanistic link between dobutamine and ROCK2 inhibition.
3.1. Dobutamine Inhibits ROCK2 Enzymatic Activity without Affecting PLK1
Drug repurposing is an attractive strategy in medicinal chemistry. While developing new drugs requires substantial investment and a long timeline, repurposing existing drugs can offer new therapeutic options using compounds that are already approved [1].
The paradigm of drug repurposing in oncology has evolved significantly in recent years, driven by advances in computational methodologies and the need for rapid therapeutic innovation. A distinctive feature of kinase inhibitors is their potential for target promiscuity, which makes them highly attractive candidates for repurposing efforts [46]. As highlighted by a recent comprehensive analysis, the promiscuous nature of kinase inhibitor binding, owing to the conserved ATP-binding pocket architecture, enables these compounds to interact with multiple kinases beyond their original intended targets, revealing unexpected therapeutic opportunities [18]. The integration of molecular docking, machine learning, and artificial intelligence has further accelerated this process, enabling computational identification of novel kinase-ligand interactions that can be rapidly validated experimentally [47].
To test our primary hypothesis, we first assessed the ability of dobutamine to directly inhibit the enzymatic activity of recombinant human ROCK2. The results demonstrated that dobutamine inhibits ROCK2 in a dose-dependent manner, yielding an IC50 of 7.1 µM (Fig. 2). The dose-response curve exhibited a Hill slope of 0.85, which is consistent with a well-behaved, non-artifactual inhibition profile [48]. This finding provides the first direct evidence mechanistically linking the cardiovascular drug dobutamine to the inhibition of the oncogenic kinase ROCK2, thereby offering a plausible molecular explanation for its previously reported anticancer activities.
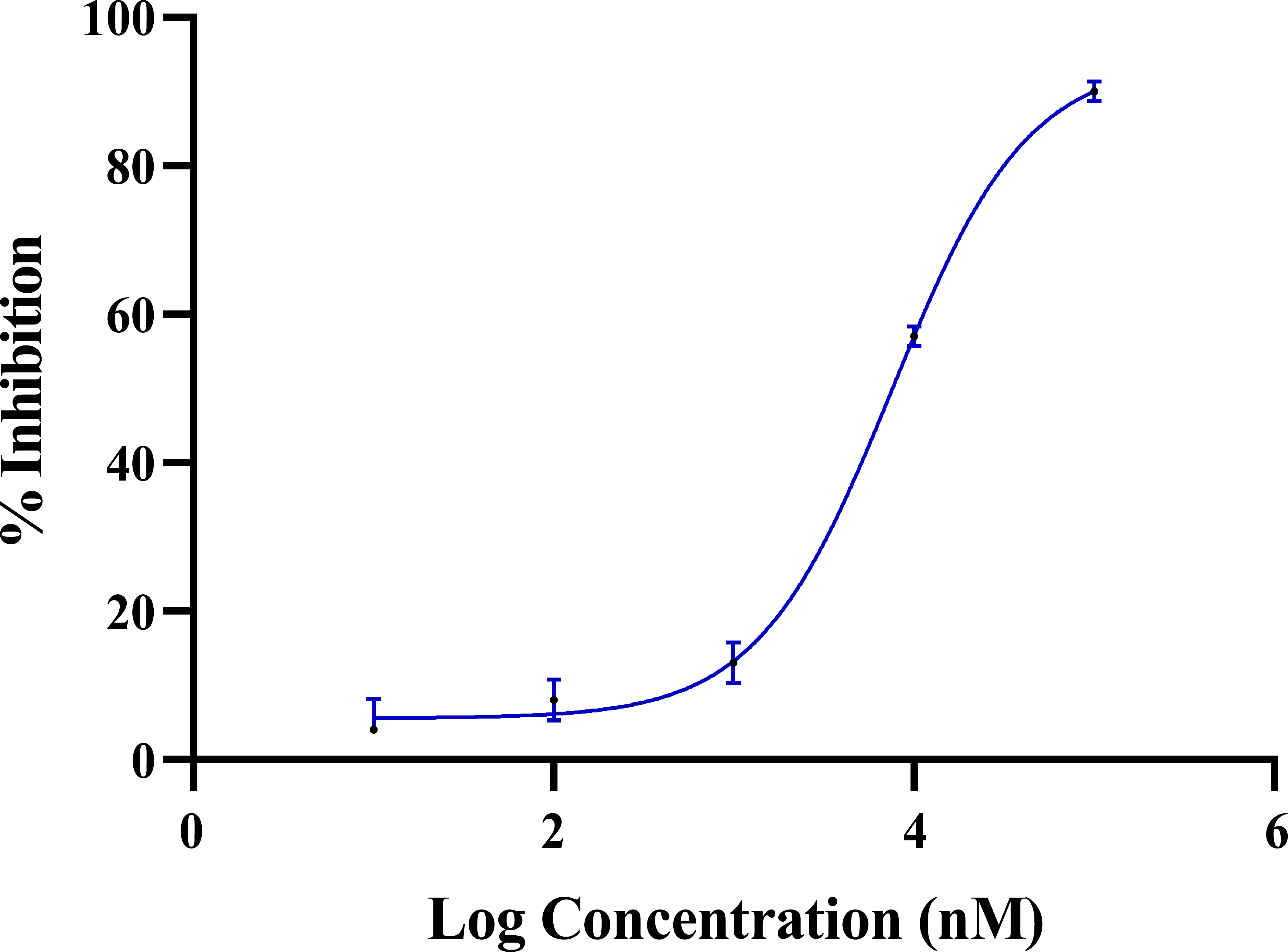
Dose-response curve of dobutamine against ROCK2. Error bars represent the standard deviation of two experimental data points.
To benchmark this result, we compared dobutamine's potency against established ROCK2 inhibitors. Hydroxyfasudil (HA-1100), a well-characterized ROCK inhibitor and active metabolite of fasudil, exhibits IC50 values of 0.72–0.73 µM for ROCK2 [49]. The pan-ROCK inhibitor Y-27632 demonstrates IC50 values of 0.14–0.22 µM for both ROCK1 and ROCK2 [50]. Ripasudil (K-115), an ophthalmic ROCK inhibitor approved in Japan, shows IC50 values of 19 nM (0.019 µM) for ROCK2 and 51 nM (0.051 µM) for ROCK1 [51]. Belumosudil (KD025), a ROCK2-selective inhibitor recently approved by the FDA for chronic graft-versus-host disease, exhibits remarkable selectivity with an IC50 of 105 nM (0.105 µM) for ROCK2 versus 24 µM for ROCK1 [52, 53]. Thus, while dobutamine's IC50 of 7.1 µM is approximately 10–100-fold weaker than these dedicated ROCK inhibitors, its confirmed activity validates the scaffold for further optimization and represents a novel chemotype for this target class.
Although an IC50 of 7.1 µM is modest potency for a lead compound, it is still significant for a repurposed drug discovered through a hypothesis-driven approach [1, 2]. This activity is comparable to dobutamine's reported off-target inhibitory activities against other kinases, such as FYN (IC50 = 3.98 µM), LCK (IC50 = 5.52 µM), and EGFR (IC50 = 5.55 µM), suggesting it may act as a multi-kinase inhibitor [54]. To probe for selectivity, we also tested dobutamine against Polo-like kinase 1 (PLK1), which can activate ROCK2. At a concentration of 10 µM, dobutamine showed only 7% inhibition of PLK1, indicating a notable degree of selectivity for ROCK2 over this related kinase.
While we observed selectivity for ROCK2 over PLK1, an important consideration is the potential cross-reactivity between ROCK2 and its closely related isoform ROCK1. These two kinases share approximately 65% overall sequence identity and 92% identity within their kinase domains [55]. Although dobutamine demonstrated a clear inhibitory activity against ROCK2 through binding to the ATP-binding site, the high degree of sequence identity between the ATP-binding domains of ROCK1 and ROCK2 (≈92%) suggests that it is likely to inhibit both isoforms with similar potency. This is consistent with the behavior of other ATP-competitive ROCK inhibitors such as Y-27632, fasudil, and hydroxyfasudil, which lack selectivity between the two isoforms. Indeed, achieving ROCK2 selectivity remains a significant challenge in drug design and typically requires targeting unique structural features outside the ATP-binding pocket or exploiting allosteric sites, as exemplified by belumosudil [52, 53]. The reported off-target activities of dobutamine against other kinases (FYN, LCK, and EGFR with IC50 values of 3.98–5.55 µM) [54] suggest a promiscuous kinase inhibition profile that may contribute to its pleiotropic cellular effects. However, it should be noted that these IC50 values are within the same micromolar range as its ROCK2 activity, and the therapeutic relevance of these interactions requires further investigation in cellular and in vivo contexts.
3.2. Dobutamine Selectively Suppresses the Proliferation of ROCK2-Overexpressing Cells
Having established that dobutamine can directly inhibit ROCK2, we next sought to determine if this molecular activity translates into a functional cellular effect. We hypothesized that if ROCK2 is a primary intracellular target, then cells with higher levels of ROCK2 expression should be more sensitive to dobutamine's antiproliferative effects. To test this, we selected two human cancer cell lines with markedly different ROCK2 expression levels: the hepatocellular carcinoma cell line Hep-G2 (high ROCK2 expression; normalized Transcripts Per Million = 85.5) and the breast ductal carcinoma cell line T-47D (low ROCK2 expression; nTPM = 8.8) [32].
The cells were treated with increasing concentrations of dobutamine for 72 hours, and cell viability was assessed using the MTT assay. As shown in Figure 3, dobutamine exhibited significantly greater antiproliferative potency against high-ROCK2-expressing Hep-G2 cells, with an IC50 of 15.2 µM (95% Confidence interval: 12.6 to 18.5). In contrast, the low-ROCK2-expressing T-47D cells were substantially less sensitive, with an IC50 of 56.1 µM (95% Confidence interval: 48.5 to 64.3). The difference between the two cell lines was statistically significant, with a p-value < 0.05, as indicated by the non-overlapping 95% confidence intervals [45]. This significant difference in potency (around 3.7 times) provides a strong correlation between cellular ROCK2 expression and sensitivity to dobutamine. This result powerfully bridges the gap between molecular target engagement and a whole-cell phenotype, providing compelling evidence that ROCK2 inhibition is a key mechanism mediating the drug's antiproliferative effects at the cellular level.
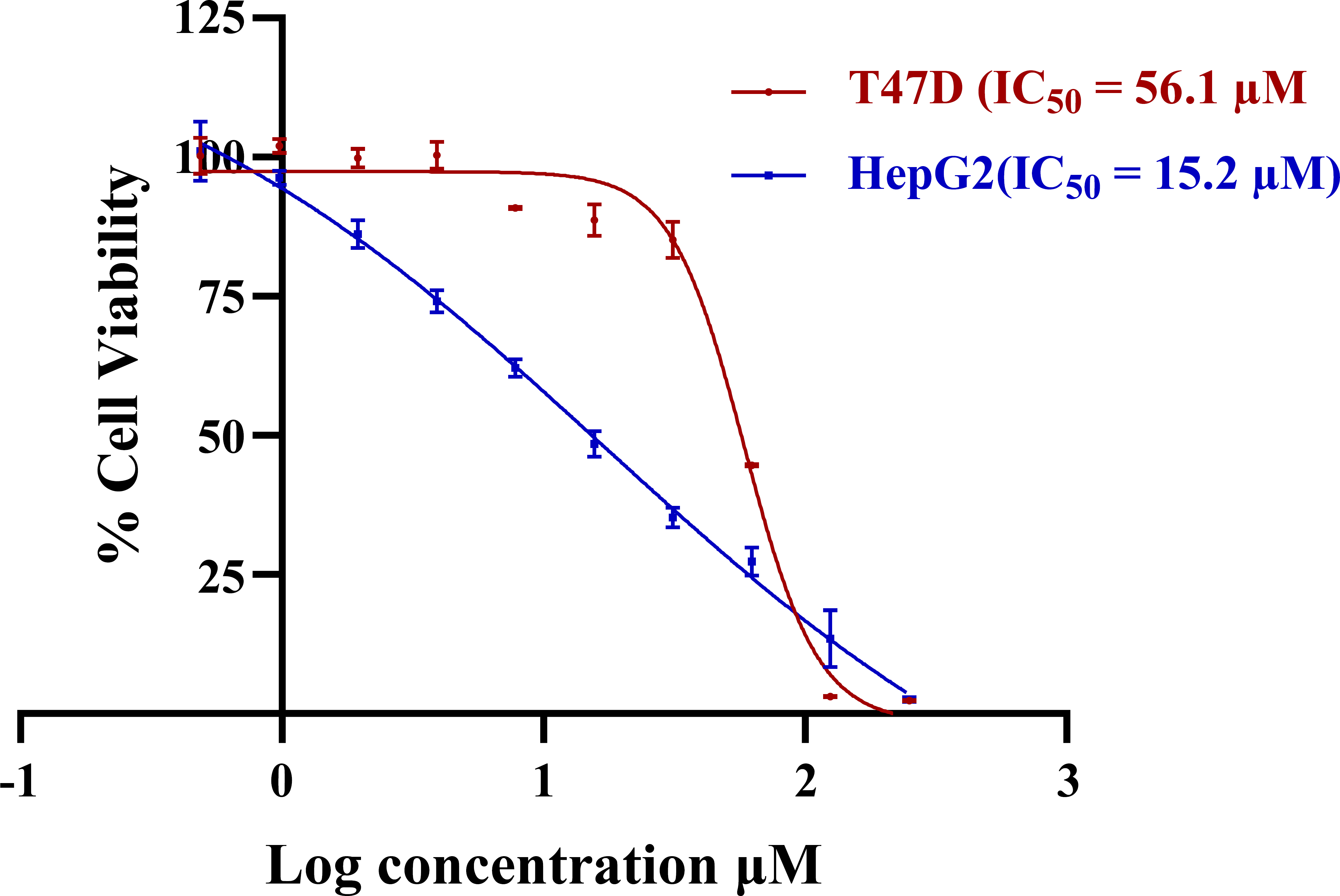
Dose-dependent cytotoxicity of dobutamine in Hep-G2 and T-47D. Error bars represent the standard deviation of experimental data points (n=4).
3.3. Molecular Docking for Dobutamine in the ROCK2 Active Site
To understand the structural basis for dobutamine's inhibition of ROCK2, we performed molecular docking studies. We used the high-resolution (2.2 Å) crystal structure of human ROCK2 (PDB ID: 8X92), which was selected due to the presence of a well-defined co-crystallized ligand in the ATP-binding site, providing a high-quality receptor model. The docking protocol was first validated via self-docking of the co-crystallized ligand (YEW), which successfully reproduced the experimental binding pose with a root mean square deviation (RMSD) of 1.27 Å, confirming the reliability of our setup (Fig. 4).
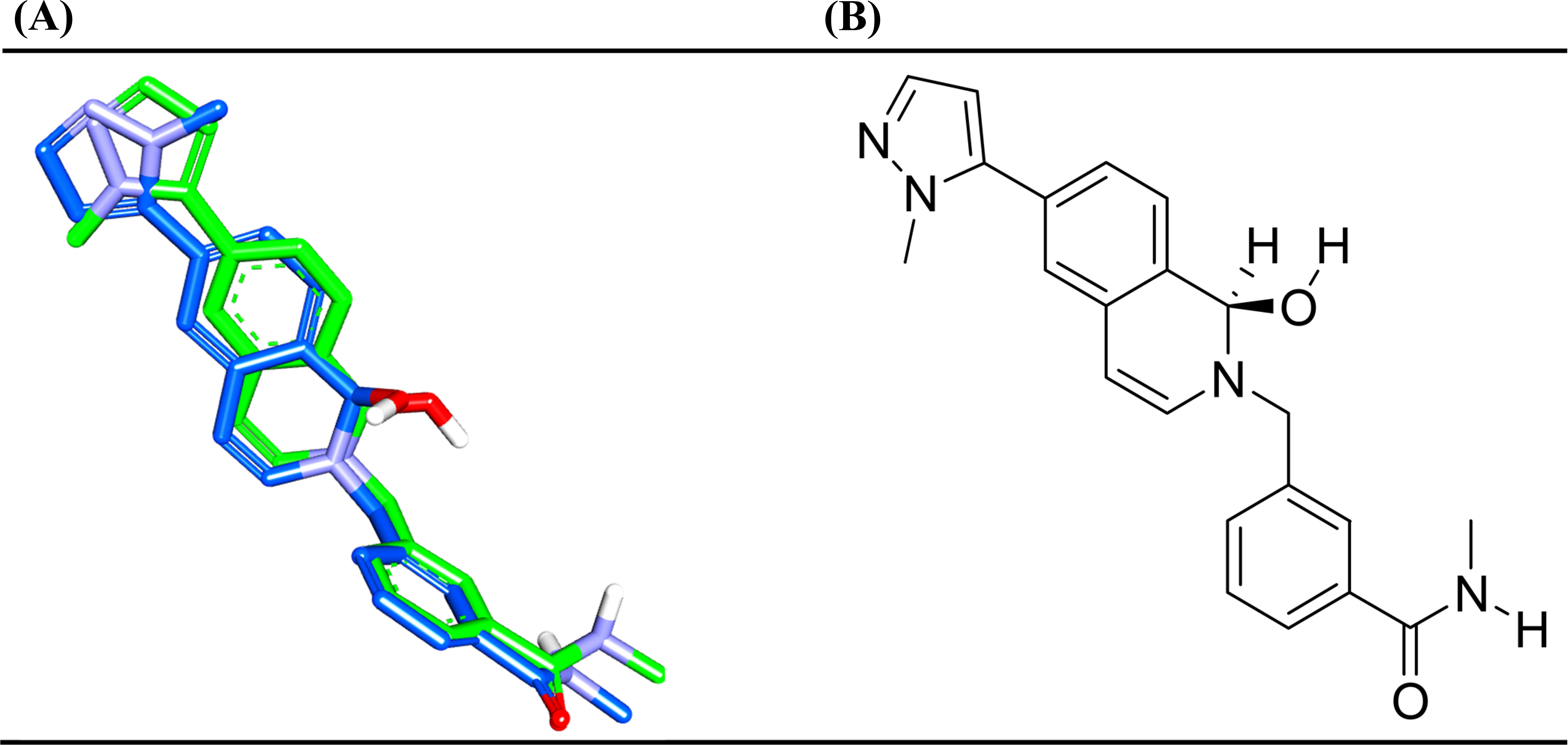
(A) High-ranking docked poses (blue) compared to crystallographic bound pose (green) of the co-crystallized ligand, YEW (PDB code: 8X92) according to PLP1, PLP2, Jain and LigScore1. (B): 2D structure of the YEW.
Dobutamine was then docked into the ROCK2 ATP-binding site. The most probable binding mode, consistently predicted by the LigScore1 and PLP1 scoring functions, is shown in (Fig. 5A and B).
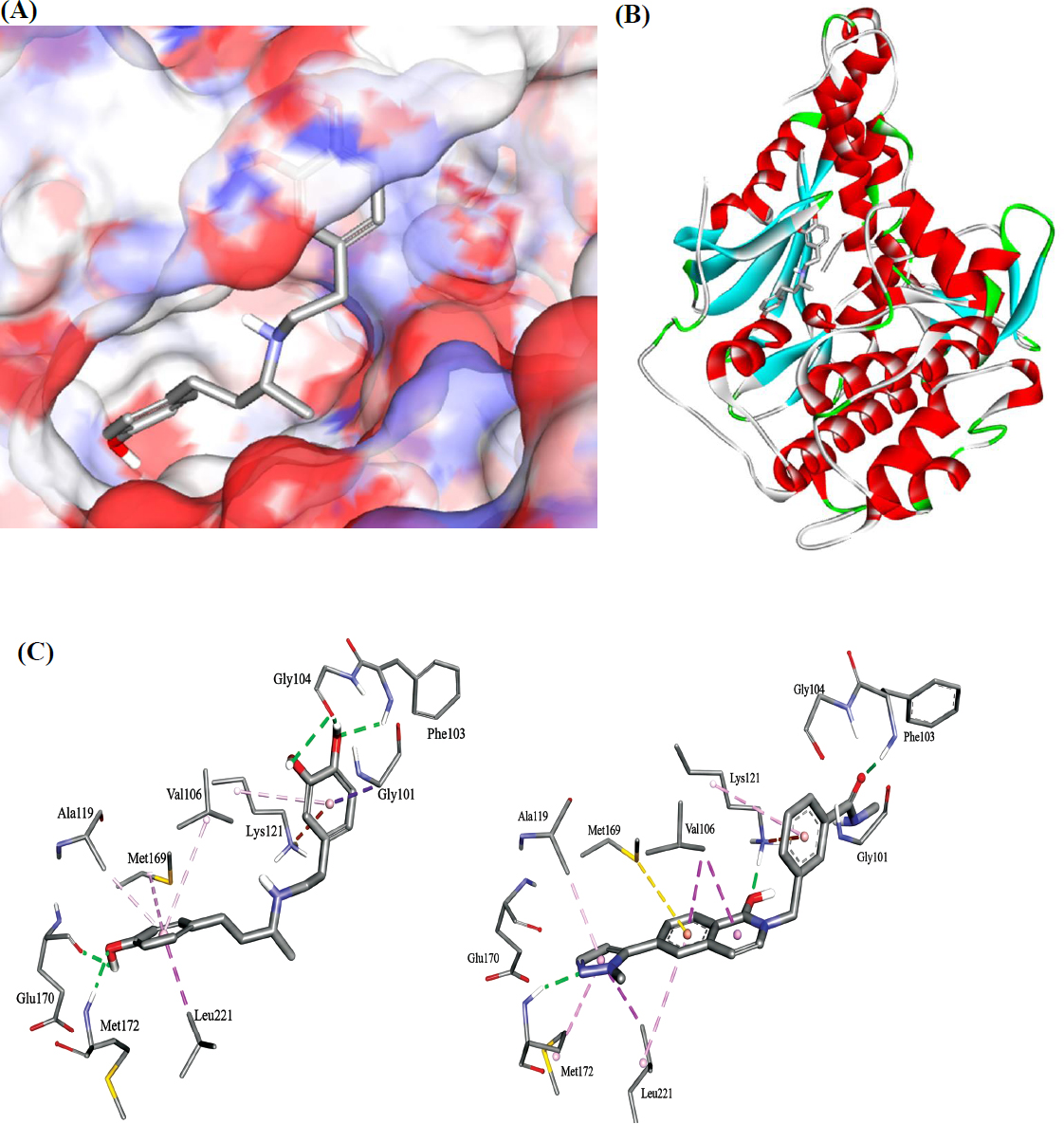
The docked pose of dobutamine in ROCK2 (PDB code: 8X92, resolution 2.2 Å). (A): Binding site covered by a solvent-accessible surface. (B): Overall view of dobutamine in the binding site. (C): Detailed view showing the binding interactions anchoring dobutamine (left) and YEW (right) within the ROCK2 kinase binding site. Hydrogen bonds, hydrophobic, and electrostatic attractive interactions are shown as green, purple and orange dotted lines, respectively.
In order to further demonstrate the reliability of the dobutamine pose, the suggested dobutamine poses were compared to the co-crystal structures of YEW based on scores from the two valid scoring functions. Docking study demonstrated that YEW produced LigScore1 of 3.28 and PLP1 of 127.05, while dobutamine produced LigScore1 of 3.38 and PLP1 of 108.25. The fact that the LigScore1s were similar indicates that both compounds, YEW and dobutamine, would likely produce similar binding affinities for ROCK2. On the other hand, the lower score of dobutamine compared to YEW using PLP1 function correlates with experimentally derived micromolar potency of dobutamine. Accordingly. PLP1 provides a strong basis for confidence in the predicted dobutamine–ROCK2 complex. Table 1 summarizes the docking results.
| Parameter | Dobutamine | YEW (8X92) | Interpretation |
|---|---|---|---|
| LigScore1a | 3.38 | 3.28 | Similar values indicate comparable shape/electrostatic complementarity |
| PLP1b | 108.25 | 127.05 | Lower score for dobutamine consistent with weaker (µM vs nM) binding |
| Self-docking RMSD | N/A | 1.27 Å | Protocol validated (threshold: <2.0 Å) |
| Experimental IC50 | 7.1 µM | nM range | ~100-fold difference in potency |
Note: aEmpirical piecewise linear potential validated for pose selection, bEmpirical piecewise linear potential optimized for binding affinity.
It is important to note that docking scoring functions are designed primarily for pose prediction and relative compound ranking rather than absolute binding affinity prediction [4]. The correlation between docking scores and experimental IC50 values is inherently limited by several well-documented factors [56, 57]. First, scoring functions employ simplified representations of protein-ligand interactions that do not fully account for entropic contributions, protein flexibility, and explicit solvent effects [58]. Second, IC50 values represent functional inhibition under specific assay conditions and are influenced by factors beyond binding affinity, including compound solubility, assay kinetics, and ATP concentration [59]. Third, scoring functions assign fixed weights to interaction terms, whereas the actual contribution of each interaction type varies in a target-dependent and non-additive manner [56]. Consequently, while our docking results provide valuable qualitative insights into the binding mode and key interactions of dobutamine within the ROCK2 active site, a direct quantitative correlation with the experimental IC50 of 7.1 µM is not expected. The comparable LigScore1 values for dobutamine (3.38) and the potent reference inhibitor YEW (3.28), together with the lower PLP1 score for dobutamine (108.25 vs. 127.05), are qualitatively consistent with dobutamine exhibiting moderate micromolar-range inhibitory activity, supporting the validity of the predicted binding pose without implying a mathematical relationship between score magnitude and potency [37, 60].
The analysis revealed that dobutamine is anchored within the kinase domain through a network of favorable interactions. Key hydrogen bonds are formed between the catechol hydroxyl groups and the backbone of residues in the critical hinge region (Gly104, Phe103), a hallmark of ATP-competitive kinase inhibitors. Additional hydrogen bonds are formed between dobutamine's phenolic hydroxyl and the side chains of Met172 and Glu170. The molecule is further stabilized by hydrophobic contacts between its aliphatic backbone and residues such as Val106, Ala119, and Leu221, as well as a favorable electrostatic (π-cation) interaction between the electron-rich catechol ring and the positively charged side chain of Lys121.
Based on the docking analysis and comparison with established ROCK inhibitor pharmacophores, the essential functional groups of dobutamine for ROCK2 inhibition can be identified [61, 62], as in Table 2. The phenol moiety represents the primary pharmacophoric element, functioning as a hinge-binding scaffold analogous to the indazole, pyrazole, and isoquinoline moieties employed by approved ROCK inhibitors such as belumosudil and fasudil [63]. This hydroxyl provides both hydrogen bond donor and acceptor capabilities essential for engaging the kinase hinge backbone, a conserved feature across ATP-competitive kinase inhibitors [64]. It hydrogen bonds with Met172 and Glu170 residues that have been identified as key determinants of ROCK2 inhibitor potency in prior SAR studies [65]. The catechol hydroxyls, on the other hand, seem to engage in hydrogen bonding with Phe103 and Gly104 of the glycine-rich loop, further stabilizing the binding interaction. In contrast, the secondary amine within the aliphatic linker, while contributing to overall molecular polarity, does not form direct hydrogen bonds with the protein and is positioned in a solvent-exposed region adjacent to the hydrophobic residue Val106. This observation suggests that the amine represents a potential site for structural modification to reduce desolvation penalties without compromising target engagement. The aliphatic chain connecting the two aromatic systems provides conformational flexibility and enables hydrophobic contacts with Val106, Ala119, and Leu221, though the entropic cost of constraining this flexible linker upon binding likely contributes to the modest micromolar potency observed [66].
| Interaction Typea | ROCK2 Residue | Protein Atom/Group | Dobutamine Functional Group | Distance (Å) | Functional Role |
|---|---|---|---|---|---|
| Hydrogen Bond | Gly104 | Backbone C=O | 3-OH (catechol) | 2.8–3.2 | (GK+1)b |
| Hydrogen Bond | Phe103 | Backbone N–H | 4-OH (catechol) | 2.8–3.2 | (GK+3) |
| Hydrogen Bond | Met172 | Side chain S | para-OH (phenyl) | 3.0–3.5 | ROCK2 specificity |
| Hydrogen Bond | Glu170 | Side chain COO− | para-OH (phenyl) | 2.6–3.0 | ROCK2 specificity |
| π-Cation | Lys121 | Side chain NH3+ | Catechol ring | 3.5–4.5 | Catalytic lysine engagement |
| Hydrophobic | Val106 | Side chain (isopropyl) | Aliphatic linker | 3.5–4.0 | Pocket filling |
| Hydrophobic | Ala119 | Side chain (methyl) | Aliphatic linker | 3.5–4.0 | Pocket filling |
| Hydrophobic | Leu221 | Side chain (isobutyl) | Aliphatic linker | 3.5–4.0 | Pocket filling |
Note: aDistance Ranges: Hydrogen bonds: 2.5–3.5 Å (heavy atom distances); π-Cation interactions: 3.5–4.5 Å (cation to ring centroid); Hydrophobic contacts: 3.5–4.0 Å (van der Waals contact distance). bNomenclature: GK = Gatekeeper residue, GK+1, GK+3 = Residues downstream from gatekeeper that form canonical hinge interactions. ATP-competitive inhibitors characteristically form 1–3 hydrogen bonds with these residues.
Table 3 and Figure 5C compare dobutamine's predicted binding interactions within the ROCK2 ATP-binding site with those of the co-crystallized ligand YEW (PDB ID: 8X92). The two compounds exhibit striking similarity in their interaction profiles, sharing hydrogen bonds with critical residue Phe103, contacts with ROCK2-specificity determinants (Met172, Glu170), and hydrophobic interactions with the adenine pocket (Val106, Ala119, Leu221). This pharmacophoric conservation validates dobutamine's predicted binding mode despite its structurally distinct catechol scaffold.
| Interaction | Dobutamine | YEW (PDB code: 8X92) |
|---|---|---|
| GK+1 / H-bond 1 | Gly104 (C=O) | No H-bond with Gly104 |
| GK+3 / H-bond 2 | Phe103 (N-H) | Phe103 (N-H) |
| Met172 contact | Yes (H-bond) | Yes (H-bond) |
| Glu170 contact | Yes (H-bond) | No (H-bond) |
| Lys121 interaction | π-cation | π-cation /H-bond |
| Hydrophobic contacts | Val106, Ala119, Leu221 | Val106, Ala119, Leu221 |
| Hinge-binding moiety | Phenol | Indazole |
| Experimental IC50 | 7.1 µM | nM (potent) |
Note: Nomenclature: GK = Gatekeeper residue, GK+1, GK+3 = Residues downstream from gatekeeper that form canonical hinge interactions. ATP-competitive inhibitors characteristically form 1–3 hydrogen bonds with these residues.
While the docked pose reveals several favorable interactions, the modest binding affinity (IC50 = 7.1 µM) can be rationalized from a structural and thermodynamic perspective. The significant conformational flexibility of dobutamine's aliphatic chain, which contains multiple rotatable bonds, incurs a substantial entropic cost upon adopting the constrained, bioactive conformation required for binding [67, 68]. Furthermore, the presence of four polar functional groups (three hydroxyls and one amine) necessitates a significant enthalpic penalty for desolvation prior to entering the relatively hydrophobic ATP-binding pocket [69, 70].
Importantly, the predicted ATP-competitive binding mode, centered on the highly conserved kinase hinge region, suggests that dobutamine is unlikely to possess significant selectivity for ROCK2 over the closely related ROCK1 isoform. The ATP-binding sites of these two kinases are nearly identical, and achieving selectivity typically requires exploiting subtle differences in peripheral regions or targeting allosteric sites. This represents a key challenge for the future development of this scaffold, as ROCK2 selectivity is a primary goal in modern inhibitor design to mitigate potential side effects associated with pan-ROCK inhibition.
3.4. Dobutamine as a Lead Scaffold for Next-Generation ROCK2 Inhibitors
The docking model not only provides a rationale for dobutamine's activity but also serves as a valuable base for structure-based drug design aimed at improving its potency and selectivity. Based on the identified structural and thermodynamic liabilities, a clear medicinal chemistry optimization strategy can be proposed.
To address the high entropic penalty associated with binding [67, 68], rigidification of the scaffold is paramount. This could be achieved by incorporating the flexible butylamine chain into a cyclic system, such as a piperidine or pyrrolidine ring. Such a modification would pre-organize the molecule into a lower-energy bioactive conformation, reducing the entropic cost of binding and potentially enhancing affinity.
To tackle the enthalpic penalty of desolvation [69, 70], modifications to reduce polarity are necessary. The docking pose (Fig. 5) indicates that the secondary amine is solvent-exposed and surrounded by lipophilic residue (Val106). As this moiety does not form key hydrogen bonds, it could be replaced with a less polar group (e.g., an ether linkage) or incorporated into a heterocyclic ring to reduce the desolvation penalty without disrupting essential interactions.
While dobutamine's potency is modest compared to dedicated, novel ROCK2 inhibitors like Zelasudil (RXC007) [23] or newly discovered pyrazolone-based scaffolds [71], its established clinical safety profile and novel chemotype for this target class make it a valuable starting point. This study successfully validates the dobutamine scaffold for ROCK2 inhibition, providing a solid foundation for future structure-based design campaigns. Such campaigns can now rationally pursue the development of potent and, crucially, selective ROCK2 inhibitors for oncologic indications.
Importantly, as an FDA-approved medication with over four decades of clinical use, dobutamine possesses a well-characterized safety and pharmacokinetic profile that can be leveraged in repurposing efforts [1]. The ROCK2-expression-dependent antiproliferative activity observed in this study-with HepG2 cells (high ROCK2 expression) exhibiting 3.7-fold greater sensitivity than T-47D cells (low ROCK2 expression)-suggests that the observed effects are target-mediated rather than indicative of indiscriminate cytotoxicity. This mechanistic selectivity, combined with dobutamine's established clinical tolerability, supports further investigation of its potential as a ROCK2-targeted anticancer agent in appropriate preclinical models.
Our findings add dobutamine to the growing list of cardiovascular drugs with demonstrated anticancer potential. This expanding class includes propranolol, which exhibits anti-metastatic effects through β-adrenergic receptor blockade and is currently in clinical trials for multiple cancer types [72]; carvedilol, which suppresses osteosarcoma and breast cancer cell viability through growth factor receptor interference [73]; and losartan, which normalizes the tumor microenvironment by reducing stromal fibrosis [74]. Unlike these agents, which primarily exert their effects through modulation of adrenergic, angiotensin, or ion channel signaling, dobutamine appears to act through direct kinase inhibition-specifically, ROCK2. This mechanistic distinction positions dobutamine as a unique addition to the cardiovascular-to-oncology repurposing paradigm and suggests that systematic screening of cardiovascular drugs against kinase panels may reveal additional repurposing opportunities.
4. STUDY LIMITATIONS
Several limitations of this study should be acknowledged. The enzymatic IC50 of 7.1 µM represents modest potency that would require significant optimization for therapeutic application. ROCK1/ROCK2 selectivity was not experimentally determined, and given the highly conserved ATP-binding sites between these isoforms (92% sequence identity within the kinase domain), dobutamine likely functions as a pan-ROCK inhibitor. The cellular studies were limited to two cancer cell lines, and broader validation across additional ROCK2-dependent cancer models is warranted. Furthermore, in vivo efficacy and pharmacokinetic considerations were not addressed, and the relative contribution of ROCK2 inhibition versus β-adrenergic receptor modulation to dobutamine's cellular effects was not deconvoluted in this study.
CONCLUSION
This study provides the first direct evidence that the cardiovascular drug dobutamine inhibits ROCK2 kinase activity, offering a mechanistic explanation for its previously reported anticancer effects. Biochemical assays demonstrated that dobutamine inhibits recombinant human ROCK2 with an IC50 of 7.1 µM and a Hill slope of 0.85, consistent with well-behaved, single-site inhibition. This molecular activity translated to a ROCK2-expression-dependent cellular phenotype, with dobutamine exhibiting 3.7-fold greater antiproliferative potency against high-ROCK2-expressing HepG2 cells (IC50 = 15.2 µM) compared to low-ROCK2-expressing T-47D cells (IC50 = 56.1 µM; p < 0.05). Molecular docking studies revealed that dobutamine occupies the ATP-binding site of ROCK2, forming hydrogen bonds with hinge-region residues (Gly104, Phe103) and ROCK2-specificity determinants (Met172, Glu170), providing a structural rationale for the observed inhibitory activity. Structure-activity relationship analysis identified the catechol moiety and para-hydroxyl group as essential pharmacophoric elements responsible for target engagement, while the secondary amine and flexible aliphatic linker were identified as structural liabilities contributing to modest potency through desolvation penalties and entropic costs, respectively.
These findings establish dobutamine as a novel chemical scaffold for ROCK2 inhibition, structurally distinct from approved inhibitors such as belumosudil and fasudil, and provide a validated binding pose that can serve as a blueprint for rational optimization campaigns. These findings are particularly timely given the growing recognition of RhoA-ROCK2 signaling as a critical driver of cancer progression and therapeutic resistance, and the expanding role of kinase-targeted therapies in oncology. This work demonstrates the utility of hypothesis-driven computational approaches in drug repurposing, particularly for kinase targets where conserved binding site architecture facilitates the identification of unexpected drug-target interactions.
Future investigations should focus on medicinal chemistry optimization to improve potency through scaffold rigidification and polarity reduction at the identified liability sites, experimental determination of ROCK1/ROCK2 selectivity, validation in additional cancer models with characterized ROCK2 dependency, and in vivo efficacy studies to assess therapeutic potential. In summary, this study establishes a novel mechanistic link between dobutamine and ROCK2 inhibition, providing both a rationale for its anticancer effects and a foundation for the development of improved ROCK2-targeted agents.
AUTHORS’ CONTRIBUTIONS
The authors confirm contribution to the paper as follows: S.D.: Conceptualization, investigation, writing; A.A.: Analysis, review & editing; R.A.: Investigation, writing; M.O.T.: Conceptualization, review & editing, supervision. All authors have read and agreed to the published version of the manuscript.
LIST OF ABBREVIATIONS
| ROCK2 | = Rho-associated Protein Kinase 2 |
| PLK1 | = Polo-Like Kinase 1 |
| FRET | = Fluorescence Resonance Energy Transfer |
| DMEM | = Dulbecco’s Modified Eagle Medium |
| SD | = Standard Deviation |
| CI | = Confidence Intervals |
AVAILABILITY OF DATA AND MATERIALS
The data and supportive information is available within the article.
FUNDING
This research was funded by the Applied Science Private University, Amman, Jordan, Grant Number: DRGS/2025/7.
ACKNOWLEDGEMENTS
The authors thank the Deanships of Scientific Research at Applied Sciences Private University (Amman) and the University of Jordan for sponsoring this project.

