All published articles of this journal are available on ScienceDirect.

Glycine Transport Inhibitors for the Treatment of Schizophrenia
Abstract
Multiple lines of evidence indicate that hypofunction of glutamatergic neurotransmission via N-methyl-D-aspartate (NMDA) receptors might be implicated in the pathophysiology of schizophrenia, suggesting that increasing NMDA receptor function via pharmacological manipulation could provide a new strategy for the management of schizophrenia. Currently, the glycine modulatory sites on NMDA receptors present the most attractive therapeutic targets for the treatment of schizophrenia. One means of enhancing NMDA receptor neurotransmission is to increase the availability of the obligatory co-agonist glycine at modulatory sites on the NMDA receptors through the inhibition of glycine transporter-1 (GlyT-1) on glial cells. Clinical studies have demonstrated that the GlyT-1 inhibitor sarcosine (N-methyl glycine) shows antipsychotic activity in patients with schizophrenia. Accordingly, a number of pharmaceutical companies have developed novel and selective GlyT-1 inhibitors for the treatment of schizophrenia. This paper provides an overview of the various GlyT-1 inhibitors and their therapeutic potential.
INTRODUCTION
A growing body of evidence suggests that the N-methyl-D-aspartate (NMDA) receptors (Fig. 1) play a role in the pathophysiology of schizophrenia [1-12]. Subanesthetic doses of the non-competitive NMDA receptor antagonist phencyclidine (PCP; Fig. 2) have been shown to produce a wide range of transient schizophrenia-like symptoms, including estrangement, loss of body boundaries, formal thought disorder, hallucinations, and psychosis [1,13,14]. In addition, PCP is known to dramatically exacerbate the symptoms of schizophrenia [1,13,14]. In a randomized, double-blind, placebo-controlled study, Krystal and co-workers [15] reported that the non-competitive NMDA receptor antagonist ketamine (Fig. 2) produced positive and negative symptoms in a dose-dependent manner. A high dose of ketamine elicited significant perceptual effects, including altered perceptions of the body, environment, and time. In another double-blind, placebo-controlled study, Malhotra et al. [16] reported that ketamine produced brief psychosis marked by thought disorder and withdrawal-retardation in healthy volunteers. Furthermore, a double-blind, crossover-design study revealed that ketamine alters mood in healthy volunteers [17]. Moreover, in a double-blind, placebo-controlled study, Lahti et al. [18] reported that ketamine induced a brief (less than 30 minutes), dose-related worsening of positive symptoms in schizophrenic patients maintained on haloperidol. In contrast, Zarate et al. [19] demonstrated the robust and rapid (within 2 h) antidepressant effects of a single dose of ketamine (0.5 mg/kg, i.v. infusion for 40 min) in treatment-resistant major depression, suggesting the role of NMDA receptors in the rapid antidepressant activity of ketamine [20-22].
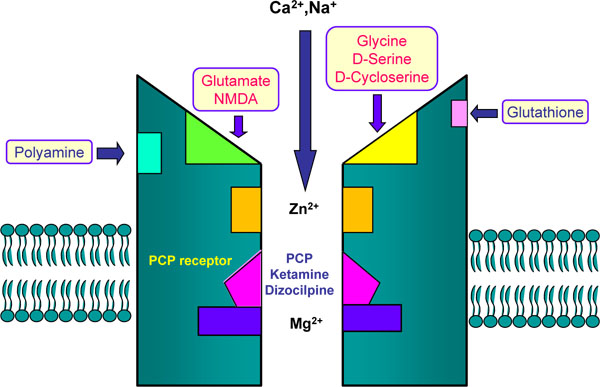
The NMDA receptor complex Glutamate and NMDA bind to the agonist site on NMDA receptors. Phencyclidine (PCP), ketamine, and dizocilpine ((+)-MK-801) bind to PCP-receptor in the inside of the NMDA receptors. Glycine and D-serine bind to co-agonist site (glycine modulatory site) on the NMDA receptors.
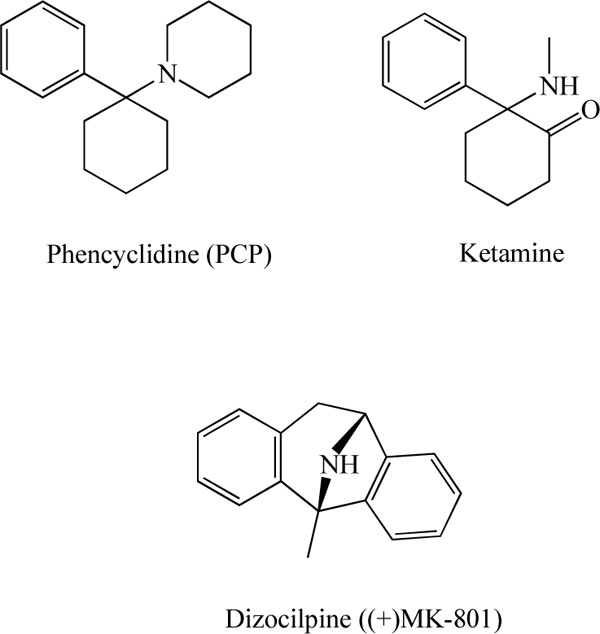
Chemical structures of phencyclidine (PCP), ketamine, and dizocilpine ((+)-MK-801).
In addition to glutamate, the NMDA receptors are modulated by glycine, D-serine, polyamines, and specific divalent cations, including magnesium and zinc [9-12] (Fig. 1). Glycine and D-serine (Fig. 3) act as obligatory co-agonists at the glycine modulatory sites on the NMDA receptors to regulate glutamatergic transmission. Currently, the glycine modulatory sites on the NMDA receptors are the most attractive therapeutic targets for schizophrenia. This review article provides an overview of glycine transporter-1 (GlyT-1) inhibitors as a potential therapeutic approach to the treatment of schizophrenia.
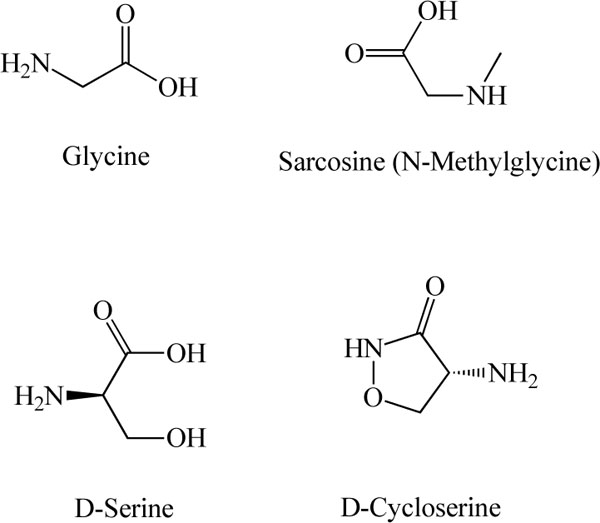
Chemical structures of glycine, sarcosine (N-methyl glycine), D-serine, and D-cycloserine
GLYCINE
Glycine (Fig. 3) was first proposed to act as a neurotransmitter in the mammalian spinal cord in 1965 [23]. Glycine is now among the well-characterized amino acid neurotransmitters in the mammalian CNS, where it is known to act as an inhibitory transmitter via its interaction with strychnine-sensitive glycine receptors [24-29]. It also plays an important role in excitatory neurotransmission via strychnine-insensitive glycine sites located on the NMDA receptors [26-29].
The plasma levels of total serine (L- and D-serine) and glycine in patients with schizophrenia are higher than those of controls [30], and the levels of serine and glycine in the brains of schizophrenic patients are higher than those of controls [30,31], suggesting a possible abnormality in serine hydroxymethyltransferase, by which glycine is converted to L-serine [10]. Interestingly, it has been reported that serum D-serine levels and the D-serine/total serine ratio in patients with schizophrenia are significantly lower than those of healthy control subjects, supporting the hypothesis of NMDA receptor hypofunction in schizophrenia [8].
Given the NMDA receptor hypofunction hypothesis of schizophrenia, increasing NMDA receptor function by glycine may be a potential strategy for the management of schizophrenia. Recent retrospective analyses have suggested that NMDA receptor agonists are effective at the treatment of persistent negative symptoms and cognitive deficits of schizophrenia, and that the full agonist glycine may be more effective than the partial agonist D-cycloserine (Fig. 3) [32].
GLYCINE TRANSPORTER (GLyT)
In the CNS, synaptic levels of glycine are regulated by specific sodium/chloride-dependent transporters. The effects of glycine in the synapse are terminated by its rapid reuptake into the nerve terminal and adjacent glial cells via high-affinity glycine transporters referred to as GlyT-1 and GlyT-2. GlyT-1 and GlyT-2 possess 12 putative transmembrane spanning domains, and share approximately 50% amino acid sequence identity [26-29,33,34]. GlyT-1 is widely expressed in the CNS, where it is predominantly present on glial cells. It is likely that GlyT-1 is responsible for glycine reuptake in forebrain areas, and in some regions, it may be co-localized with strychnine-insensitive glycine modulatory sites on the NMDA receptors [33-39]. A recent study demonstrated that glycine transport might maintain local synaptic glycine at very low levels, suggesting that GlyT-1 could play a role in regulating glutamatergic neurotransmission via NMDA receptors [40]. In contrast to GlyT-1, GlyT-2 has a predominantly neuronal and more limited distribution, being primarily restricted to the spinal cord, brainstem, and cerebellum [38]. Indeed, GlyT-2 is known to be co-localized with strychnine-sensitive glycine receptors, suggesting that GlyT-2 may be a reliable marker for glycinergic neurons [26-29].
In an attempt to clarify the in vivo functional roles of glycine transporters in the CNS, knockout mice deficient in the GlyT-1 gene have been generated [41,42]. Newborn mice deficient in the GlyT-1 gene are anatomically normal, but show severe motor and respiratory deficits and die during the first postnatal day [41]. Glycine or the GlyT-1 inhibitor sarcosine (N-methyl glycine; Fig. 3) were found to suppress respiratory activity in slices from wild-type mice. During early postnatal life, GlyT-1 is essential for regulating glycine levels at inhibitory glycine receptors, and GlyT-1 deletion generates symptoms found in human glycine encephalopathy [41]. Furthermore, heterozygous knockout mice (reduced expression of GlyT-1) show enhanced hippocampal NMDA receptor function and memory retention, and are protected against disruptions of sensory gating induced by amphetamine, which suggests that GlyT-1 inhibitors might bring about both cognitive enhancement and antipsychotic effects [42]. Mice heterozygous for the GlyT-1 gene show faster decay kinetics, reduced ifenprodil sensitivity, and increased zinc-induced antagonism in NMDA receptor currents [42]. These findings highlight the importance of GlyT-1 in the regulation of glutamatergic neurotransmission. Taken together, the data suggest that increasing synaptic levels of glycine by inhibition of its uptake will lead to enhanced NMDA receptor activation, in turn suggesting a potential role for GlyT-1 inhibitors as a novel treatment for schizophrenia [43-55].
GLyT-1 INHIBITORS
Glycine is an endogenous compound that is actively metabolized and sequestered in the brain. Glycine is extensively metabolized in the liver, and only poorly crosses the blood-brain barrier. Therefore, a large amount of glycine is needed for the treatment of schizophrenia [32]. Thus, the ability of glycine levels to modulate NMDA receptor-mediated neurotransmission suggests that the pharmacological manipulation of synaptic glycine might be effective in the treatment of conditions involving NMDA receptor hypofunction [1,43-55].
The NMDA receptor antagonist PCP (Fig. 2) has been widely used in studies of animal models of schizophrenia [9-11,56,57]. We previously reported that repeated administration of PCP (10 mg/kg/day for 10 days) caused long-term cognitive deficits in mice (more than 6 weeks after the final administration of PCP), and that PCP-induced cognitive deficits could be improved by subsequent subchronic (2 weeks) administration of clozapine, but not haloperidol [58]. Therefore, the reversal of PCP-induced cognitive deficits may be a potential animal model of atypical antipsychotic activity, i.e., this model may be beneficial in terms of ameliorating the cognitive deficits in people with schizophrenia [59-67]. Using this model, we reported that treatment with the selective GlyT-1 inhibitor, (R)-(N-[3-(4'-fluorophenyl)-3-(4'-phenylphenoxy)propyl])sarcosine (NFPS, ALX 5407; Fig. 4) [68-71], attenuated PCP-induced cognitive deficits in mice [64]. We also found that repeated administration of PCP caused increased levels of GlyT-1 protein in the mouse hippocampus, and that extracellular glycine levels in the hippocampus of PCP-treated mice were lower than those of control mice [64]. Furthermore, NFPS inhibited PCP-induced hyperactivity in mice, and NFPS reversed PCP-induced changes in electroencephalogram (EEG) power spectra in conscious rats [71]. It has been reported that NFPS induced a pattern of c-Fos immunoreactivity comparable with the atypical antipsychotic drug clozapine, and that NFPS enhanced prepulse inhibition (PPI) of the acoustic startle response in DBA/2J mice, a strain with low basal levels of PPI [72]. Moreover, to the same degree as by clozapine or D-serine, NFPS was able to reverse persistent latent inhibition (LI) [72] and cognitive deficits [73] induced by the NMDA receptor antagonist dizocilpine ((+)-MK-801; Fig. 2). Manahan-Vaughan et al. [74] reported that NFPS could rescue hippocampal long-term potentiation and learning deficits in freely behaving rats after systemic administration of dizocilpine. All these findings suggest that GlyT-1 inhibitors could potentially serve as therapeutic drugs to treat the cognitive deficits associated with schizophrenia.
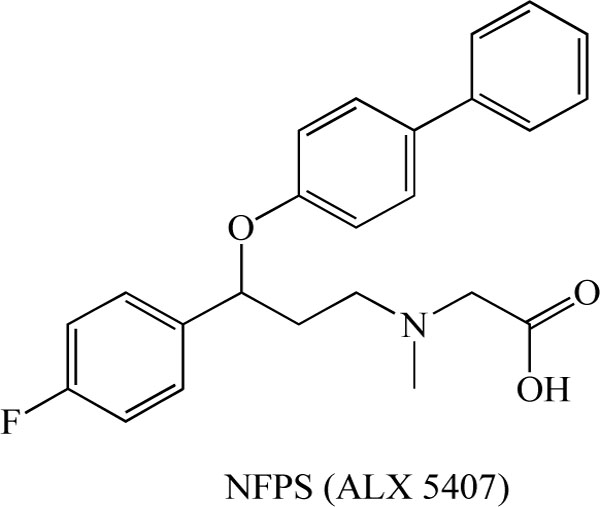
Chemical structure of (R)-(N-[3-(4'-fluorophenyl)-3-(4'- phenylphenoxy)propyl])sarcosine (NFPS, ALX 5407).
Researchers at Sanofi-Synthelabo Recherche reported the detailed neuropharmacological profile of SSR 504734 (Fig. 5) as part of a biochemical approach to developing a selective and reversible GlyT-1 inhibitor [75]. SSR 504734, a selective and reversible inhibitor of GlyT-1, blocked the ex vivo uptake of glycine in a rapid, reversible, and long-term manner. In animal models of schizophrenia, this compound normalized spontaneous PPI deficits in DBA/2 mice, and reversed both amphetamine-induced locomotor hyperactivity as well as selective attention deficits in adult rats treated neonatally with PCP. These findings suggest that SSR 504734 is a potent and selective GlyT-1 inhibitor that exhibits ameliorative effects in animal models of schizophrenia; this compound may therefore be efficacious not only in treating positive, but also negative symptoms (i.e., cognitive deficits) of schizophrenia [75]. Moreover, it has been reported that SSR 504734 (10 mg/kg) enhanced the facilitatory influence of glutamatergic afferents on dopamine neurotransmission in the nucleus accumbens, and this synergistic effect was found to be dependent on glutamatergic tone [76]. Furthermore, SSR 504734 is reported to be effective in the PCP-induced functional activation in the cortico-limbo-thalamic circuits [77] and working memory deficits [78]. Moreover, SSR 504734 attenuated PCP-induced hyperlocomotion in mice, but potentiated the motor stimulant and motor depressant effects of amphetamine and apomorphine, respectively [79].
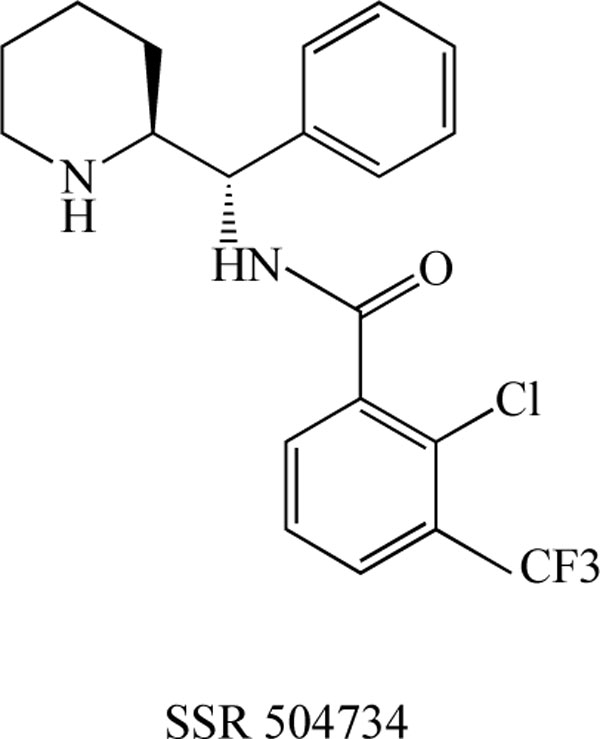
Chemical structure of SSR 504734
Recently, researchers at the Sanofi-Synthelabo Recherche Institute reported the detailed neuropharmacological profile of SSR 103800, a novel selective and reversible GlyT-1 inhibitor. They demonstrated that SSR 103800 elevates central glycine levels in the prefrontal cortex, and it exhibits potential therapeutic activity in animal models considered representative of the positive, cognitive, and depressive symptoms observed in patients with schizophrenia [80]. SSR 103800 (1 and 3 mg/kg) and SSR 504734 (1 and 10 mg/kg) potentiated latent inhibition (LI) under conditions where LI was not present in non-treated controls and SSR 103800 (1 mg/kg) reversed amphetamine-induced disrupted LI while not affecting LI on its own. Additionally, SSR 103800 (1 and 3 mg/kg) and SSR 504734 (3 and 10 mg/kg) reversed abnormally persistent LI induced by dizocilpine. In the neurodevelopmental model, SSR 504734 (3 and 10 mg/kg) reverted the LI back to control (normal) levels [78]. These preclinical data from acute and neurodevelopmental models suggest that GlyT-1 inhibitors could exhibit activity in the positive, negative, and cognitive symptom domains of schizophrenia.
Researchers at Merck Research Laboratories reported the pharmacological profile of a class of novel GlyT-1 inhibitors related to 4,4-disubstituted piperidines, including 2-methoxy-N-{1-[4-phenyl-1-(propylsulfonyl)piperidin-4-yl]-methyl}benzamide (compound 1: Fig. 6) and 2-amino-6-chloro-N-{(1S)-1-[4-phenyl-1-(propylsulfonyl)-piperidin-4-yl]ethyl}benzamide (compound 2: Fig. 6) [81,82]. A rapid and sustained increase in the extracellular levels of glycine in the prefrontal cortex of freely moving rats was observed at all three experimental doses (1, 3, 10 mg/kg, s.c.) of this compound. Furthermore, this compound was found to significantly enhance PPI at three different doses (3, 30, 100 mg/kg, s.c.) in DBA/2J mice. Brain levels of this compound ranged from 400 nM to 2300 nM during the time course of the PPI experiments. By selectively increasing extracellular glycine levels in the prefrontal cortex via the inhibition of GlyT-1, this compound significantly enhanced performance in a behavioral animal model of sensorimotor gating [81].
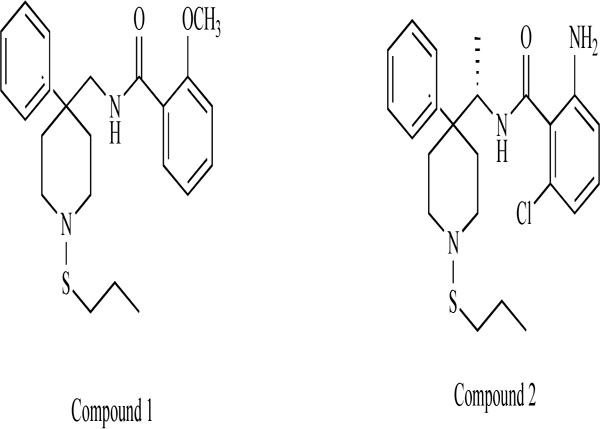
Chemical structure of compound 1 (2-methoxy-N-{[4-phenyl-1-(propylthio)piperidin-4-yl]methyl}benzamide) and compound 2 (2- amino-6-chloro-N-{(1S)-1-[4-phenyl-1-(propylthio)piperidin-4-yl]ethyl}benzamide).
Researchers at F. Hoffmann-La Roche, Ltd. reported a novel series of N-(2-aryl-cyclohexyl) substituted spiropiperidines (compound 3-7: Fig. 7) as highly selective GlyT-1 inhibitors [83-87]. Furthermore, a potent and selective GlyT-1 inhibitor 4-(4-{[2-(cyclopropylmethoxy)-5-(methylsulfonyl)phenyl]carbonyl}piperazin-1-yl)-3-fluorobenzonitrile (compound 8: EC50 = 16 nM for GlyT-1)(Fig. 8) increased extracellular glycine levels in the mouse striatum after oral administration [88].
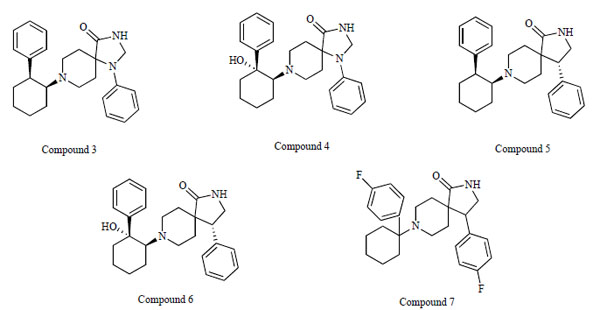
Chemical structures of compound 3, (1-phenyl-8-[(1S,2S)-2-phenylcyclohexyl]-1,3,8-triazaspiro[4.5]decan-4-one), compound 4, (8- [(1S,2R)-2-hydroxy-2-phenylcyclohexyl]-1-phenyl-1,3,8-triazaspiro[4.5]decan-4-one), compound 5, ((4R)-4-phenyl-8-[(1S,2S)-2- phenylcyclohexyl]-2,8-diazaspiro[4.5]decan-1-one), compound 6, ((4R)-8-[(1S,2R)-2-hydroxy-2-phenylcyclohexyl]-4-phenyl-2,8- diazaspiro[4.5]decan-1-one), and compound 7, (4-(4-fluorophenyl)-8-[1-(4-fluorophenyl)cyclohexyl]-2,8-diazaspiro[4.5]decan-1-one).
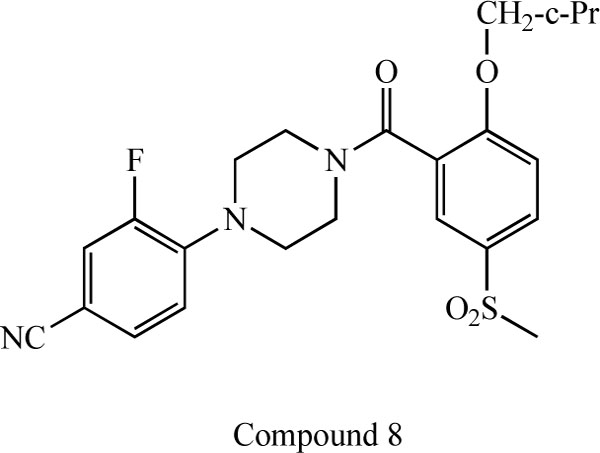
Chemical structure of compound 8, 4-(4-{[2- (cyclopropylmethoxy)-5-(methylsulfonyl)phenyl]carbonyl}piperazin- 1-yl)-3-fluorobenzonitrile.
Researchers at H. Lundbeck A/S developed the compound (R)-4-[5-chloro-2-(4-methoxy-phenylsulfanyl)-phenyl]-2-methyl-piperazin-1-yl-acetic acid (compound 9: IC50 = 150 nM) (Fig. 9), which was shown to elevate extracellular glycine levels in the rat ventral hippocampus, as measured by in vivo microdialysis at doses of 1.2-4.6 mg/kg (s.c.) [89]. Furthermore, the same group reported the new compound (S)-1-{2-[3-(3-fluoro-phenylsulfanyl)biphenyl-4-yloxy]ethyl}pyrrolidine-2-carboxylic acid (compound 10: IC50 = 59 nM) [90] (Fig. 9). In vitro and in vivo assessments revealed that the CNS utility of this class of compounds might be diminished due to active efflux transporter activity [90].
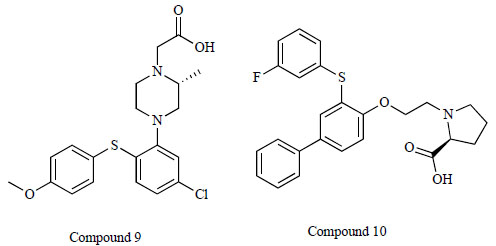
Chemical structure of compound 9, (R)-4-[5-chloro-2-(4- methoxy-phenylsulfanyl)-phenyl]-2-methyl-piperazin-1-yl-acetic acid and compound 10, (S)-1-{2-[3-(3-fluoro-phenylsulfanyl) biphenyl-4-yloxy]ethyl}pyrrolidine-2-carboxylic acid.
ORG 25935 (cis-N-methyl-N-(6-methoxy-1-phenyl-1,2,3,4-tetrahydronaphthalen-2- ylmethyl)amino-methylcarboxylic acid hydrochloride)( Fig. 10) is a GlyT-1 inhibitor that easily crosses the blood-brain barrier. ORG 25935 (6 mg/kg, i.p.) was shown to increase striatal extracellular glycine levels by ~50-80% for ~2.5 hours [91]. Molander et al. [92] reported that ORG 25935 decreased ethanol intake and ethanol preference, as compared with vehicle, without affecting water intake. This effect was dose-dependent, developed gradually, and was sustained for up to 40 days, even after the introduction of an alcohol-deprivation period. The findings suggest that ORG 25935, and possibly other GlyT-1 inhibitors as well, could provide a new pharmacological treatment strategy for alcohol dependence or abuse [92].
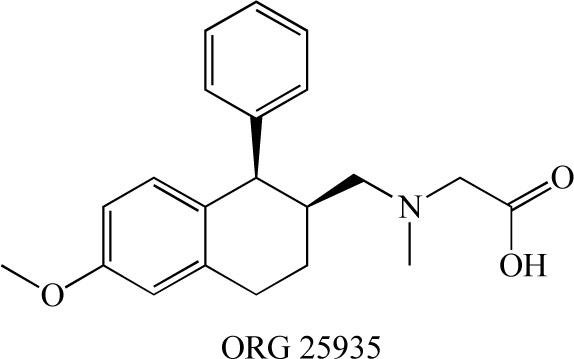
Chemical structure of ORG 25935.
BRAIN IMAGING OF GLyT-1 IN THE BRAIN
The distribution, density, and activity of GlyT-1 in the living human brain can be visualized noninvasively by the specific radioligands and positron emission tomography (PET), and single photon computed tomography (SPECT), and the receptor binding can be quantified by appropriate tracer kinetic models, which can be modified and simplified for particular applications. Therefore, in vivo PET/SPECT imaging of GlyT-1 in the human brain provides a method for quantitative study of the GlyT-1-related pathophysiology in schizophrenia. Researchers at Merck developed the novel radioligand [35S](S)-2-amino-4-chloro-N-(1-(4-phenyl-1-(propylsulfonyl)piperidin-4-yl]ethyl)benzamide ([35S]AC-PPB: Kd=1.9 nM) (Fig. 11) for GlyT-1 in the brain. Autoradiographic studies of rat and rhesus brain slices with [35S]ACPPB showed that specific binding sites were plentiful and nonhomogeneously distributed, with high levels of binding in the brainstem, cerebellar white matter, thalamus, cortical white matter and spinal cord gray matter. In vivo studies demonstrated displaceable binding of [35S]ACPPB in rat brain tissues following intravenous administration of this radioligand [93]. Investigators at Merck also developed the novel PET ligand [18F] 2,4-dichloro-N- ((1-(propylsulfonyl)-4-(6-fluoropyridin-2-yl) piperidin-4-yl)methyl)benzamide (CFPyPB) (Fig. 11) for GlyT-1 in the human brain [94].
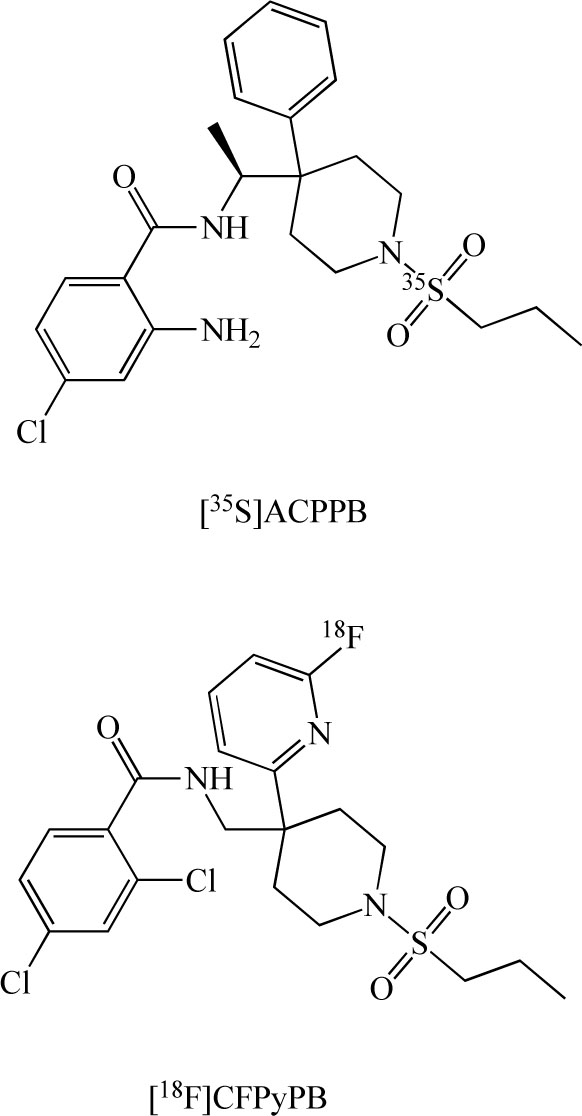
Chemical structures of [35S](S)-2-amino-4-chloro-N-(1- (4-phenyl-1-(propylsulfonyl)piperidin-4-yl)ethyl)benzamide ([35S] ACPPB), and [18F] 2,4-dichloro-N- ((1-(propylsulfonyl)-4-(6- fluoropyridin-2-yl) piperidin-4-yl)methyl)benzamide ([18F]CFPy PB).
In addition, researchers at Glaxo developed the PET ligand [11C]GSK 931145 (Fig. 12) for GlyT-1 in the human brain [95,96]. [18F]CFPyPB and [11C]GSK 931145 would be potential PET ligands for in vivo visualization of GlyT-1 in the living human brain with PET. These PET ligands represent a new tool for the evaluation of glutamatergic neurotransmission in the pathophysiology of neuropsychiatric diseases, including schizophrenia.
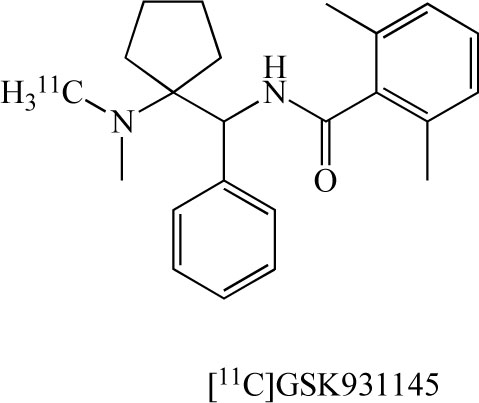
Chemical structure of [11C]GSK 931145.
CLINICAL STUDY OF GLyT-1 INHIBITORS
Sarcosine is generated by the enzymatic transfer of a methyl group from S-adenosylmethionine to glycine, and this reaction is catalyzed by the enzyme glycine N-methyltransferase [97,98]. Recently, sarcosine was identified as a biomarker that is highly increased during prostate cancer progression to metastasis and can be detected non-invasively in urine [99]. Interestingly, a recent population-based, nested, case-controlled study demonstrated a 40.7% lower risk of prostate cancer in male patients with schizophrenia [100]. It is thus likely that the sarcosine pathway plays a role in the lower risk for prostate cancer in male subjects with schizophrenia, although further detailed studies will be necessary to confirm this hypothesis [98].
Sarcosine is a safe drug, since it is synthesized in the human body. In fact, treatment with sarcosine (2 g/day) can benefit schizophrenic patients also being treated with antipsychotics, including risperidone [101], but not clozapine [102]. A randomized, double-blind, placebo-controlled study demonstrated that members of a sarcosine (2 g/day) group showed greater reductions in their Positive and Negative Syndrome Scale (PANSS) total scores than members of a placebo group or D-serine (2 g/day) group, suggesting that sarcosine is superior to D-serine in that benefits both patients with long-term stable disease and also acutely ill persons with schizophrenia [103]. Furthermore, a randomized, double-blind study reported that sarcosine (2 g/day) alone was effective in the treatment of acutely symptomatic drug-free patients with schizophrenia [104]. Further large-sized, placebo-controlled, dose-finding studies are needed to fully assess the effects of sarcosine, since these reports regarding the beneficial effects of sarcosine were based on small studies from the same group in Taiwan. In contrast, Zhang et al. [105] reported that sarcosine is an NMDA receptor co-agonist that differs from glycine, suggesting that co-agonistic activity at the NMDA receptors of sarcosine may, in part, be involved in the clinical benefits of this drug in schizophrenia. Nonetheless, all these findings suggest that GlyT-1 inhibition could be a novel pharmacotherapeutic target for enhancing NMDA receptor function.
Very recently, Liem-Moolenaar et al. [106] reported the effects of the GlyT-1 inhibitor R 231857 (Fig. 13) on the CNS and on scopolamine-induced impairments in cognitive and psychomotor function in healthy subjects. R231857 had some small effects on scopolamine-induced CNS-impairment, which were also not clearly dependent on dose. Scopolamine proved to be an accurate, reproducible and safe model for the induction of CNS impairment by an anticholinergic mechanism. R231857 lacked consistent dose-related effects in this study, probably because the CNS concentrations were too low to produce significant/reproducible CNS effects or to affect the scopolamine challenge in healthy volunteers. The effects of higher doses in healthy volunteers and the clinical efficacy in patients remain to be established. To date, clinical findings with GlyT-1 inhibitors are limited because of the early stage of development of the most high-affinity compounds.
Chemical structure of R231857.
CONCLUDING REMARKS
As described above, the glycine modulatory sites on NMDA receptors are among the most attractive targets for developing potential therapeutic drugs for the treatment of schizophrenia. One means of enhancing NMDA receptor function would be to pharmacologically increase synaptic glycine levels by GlyT-1 inhibitors. At present, a number of pharmaceutical companies are developing novel and selective GlyT-1 inhibitors for the treatment of schizophrenia [43-55]. Gaining a better understanding of the role of GlyT-1 in the treatment of schizophrenia is expected to provide new perspectives for treating this disorder. Finally, the pharmacological modulation of glycine modulatory sites on NMDA receptors by non-sarcosine-derived GlyT-1 inhibitors could be beneficial in the treatment of the cognitive deficits and psychosis associated with several psychiatric diseases, including schizophrenia.
ACKNOWLEDGEMENTS
This study was supported in part by a grant from the Minister of Education, Culture, Sports, Science, and Technology of Japan (to K.H.), and by a grant from the Program for Promotion of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation of Japan (to K.H.).

