All published articles of this journal are available on ScienceDirect.

HIV Protease Inhibitors: Effect on the Opportunistic Protozoan Parasites
Abstract
The impact of highly active antiretroviral therapy (HAART) in the natural history of AIDS disease has been allowed to prolong the survival of people with HIV infection, particularly whose with increased HIV viral load. Additionally, the antiretroviral therapy could exert a certain degree of protection against parasitic diseases. A number of studies have been evidenced a decrease in the incidence of opportunistic parasitic infections in the era of HAART. Although these changes have been attributed to the restoration of cell-mediated immunity, induced by either non-nucleoside reverse transcriptase inhibitors or HIV protease inhibitors, in combination with at least two nucleoside reverse transcriptase inhibitors included in HAART, there are evidence that the control of these parasitic infections in HIV-positive persons under HAART, is also induced by the inhibition of the proteases of the parasites. This review focuses on the principal available data related with therapeutic HIV-protease inhibitors and their in vitro and in vivo effects on the opportunistic protozoan parasites.
1. INTRODUCTION
Since the introduction of highly active antiretroviral therapy (HAART) the mortality and morbidity for a wide variety of opportunistic viral, bacterial, fungal and parasitic infections have decreased dramatically among HIV-infected individuals in economically developed countries [1].
In a multinational cohort study involving 6941 individuals with the human immunodeficiency virus (HIV) from Australia and ten European countries, when comparing the periods 1997-2001 and 1994-1996, there was a significant HAART-induced decreases (e.g. candiadiasis from 17.0% to 5.7%; cryptosporidiosis from 3.1% to 0.2%; cytomegalovirosis from 5.9% to 0.6%; Pneumocystis pneumonia from 17.0% to 4.4%; toxoplasmosis from 3.4% to 1.4% and tuberculosis from 6.4% to 2.6%) [2]. The reduction observed, when comparing both periods before HAART to the HAART era, evidence the impact of the therapy against HIV and the principal opportunistic infections associated.
The drugs used as antiretroviral therapy have been classified in six categories: nucleoside reverse transcriptase inhibitors, nucleotide reverse transcriptase inhibitors, non-nucleoside reverse transcriptase inhibitors, cell entry inhibitors (fusion inhibitors and co-receptor inhibitors), integrase inhibitors and protease inhibitors (PIs). These compounds should be used in drug combination regimens to achieve the highest possible benefit, tolerability and compliance and to diminish the risk of resistance development [3]. Although the HAART-induced reconstitution of cellular immunity seems to be the main determinant in reducing opportunistic infections in HIV-positive individuals, clinical and microbiological evidence showed that this class of drugs to be effective as antitumor, antibacterials, antifungals and anti-influenza agents [4, 5].
Even though, the target of these inhibitors is the HIV-1 protease, a member of the aspartic protease family [6], the preliminary investigations in term of action against opportunistic parasites, indicate that proteases of some parasites could be an unspecific target for HIV-1 PIs. Additionally, the results of in vitro and in vivo investigations [7], indicate that HIV-1 PIs have a direct effect on some opportunistic parasites based on aspartic protease inhibitors [5]. This article reviews general pharmacology data of PIs and summarize the relevant evidences of their effect against the principal opportunistic parasitic infections caused by Plasmodium falciparum, Leishmania spp, Toxoplasma gondii and vaginal or intestinal protozoan.
2. HIV-1 PIs
There are, at present, 25 compounds which have been formally approved by the US Food and Drug Administration (FDA) for clinical use in the treatment of the acquired immunodeficiency syndrome (AIDS), including 10 PIs (Fig. 1): saquinavir1, ritonavir2, indinavir3, nelfinavir4, amprenavir5, lopinavir6, fosamprenavir7, atazanavir8, tipranavir9 and darunavir10 [3]. PIs are small peptidic or non-peptidic proteins designed to compete for active sites of the protease enzyme. Three generation of PIs have been developed with the aim to improve the drug efficacy and/or properties such as better patient quality of live, increased survival, slowed diseases progression, decreased viral load, increased immunological response and decreased opportunist infections [8]. With the exception of tipranavir (which is based on a coumarin scaffold), all these PIs are based on the ‘peptidomimetic’ principle, that is they contain a hydroxyethylene scaffold which mimics the normal peptide linkage (cleaved by the HIV protease) [3].
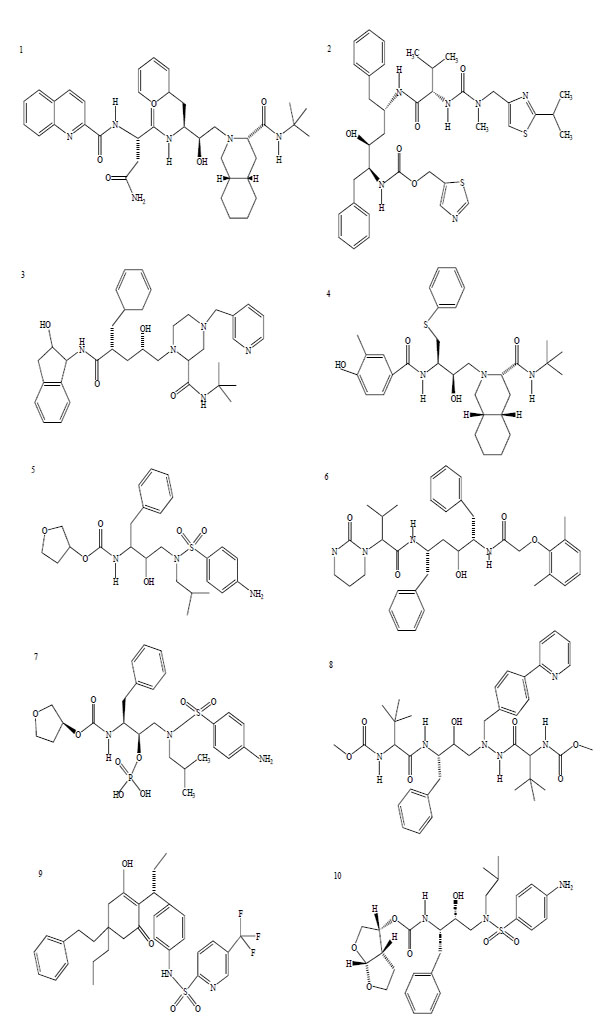
Chemical structure of HIV-1 protease inhibitors. 1: Saquinavir; 2: Ritonavir; 3: Indinavir; 4: Nelfinavir; 5: Amprenavir; 6: Lopinavir; 7: Fosamprenavir; 8: Atazanavir; 9: Tipranavir; 10: Darunavir.
To date, most treatment guidelines indicate that regimens based on lopinavir-ritonavir as the first-line agent, followed by fosamprenavir or atazanavir booster with ritonavir or as an nonboosted regimen are among the preferred options to start HAART [9].
Most PIs alone are only moderately absorbed in the gastrointestinal tract and their diffusion though anatomical barrier is usually moderate, in part due to high plasma protein binding (90-99%), which make PIs concentration lower in sanctuary sites such as the genital tract [10,11] and the CNS [12] in comparison with plasma. Compared with other PIs, fosamprenavir, lopinavir and darunavir possess better diffusion to sanctuary sites [13,14]. Food contributes to the pharmacokinetic variability of PIs, but the exact mechanism(s) are poorly understood [15]. The first advance was to increase the level of existing PIs in the plasma by boosting with ritonavir [16], which to enhance the effectiveness of PIs treatment by reducing the pill burden, simplifying dosing regimens and improving therapy adherence. Ritonavir coadministration improves the pharmacokinetic profiles of concomitant PIs, and represents a cornerstone of PI-containing regimens [17].
In term of adverse events, PIs show an acceptable safety profile. The most common short-term adverse events are gastrointestinal order, namely diarrhea [18]. Lipodystrophy is a condition associated with long-term exposure to PIs, mainly the first-generation compounds. In addition to morphological abnormalities (fat atrophy and fat deposition), metabolic disturbances (hyperglycaemia and hyperlipidemia) are common [19].
2.1. First-Generation of PIs
The fist PIs was saquinavir (Invirase® and Fortovase®) in 1995, followed in 1996 by the introduction of ritonavir (Norvir®) and indinavir (Crixivan®), in 1997 the nelfinavir (Viracept®) and in 1999 the amprenavir (Agenerase® and Prozei®) was approved. In terms of evolution of PIs in the HAART therapy, these five agents represent the first generation and at the end of the 1990s were the most widely PIs used [8].
Saquinavir is a peptidomimetic inhibitor of HIV protease with greatly hindered by its nearly complete first pass metabolism by cytochrome P450 [20]. The drug inhibits the cleavage of the gag-pol protein substrate leading to the release of structurally defective and functionally inactive viral particles. It is active on both HIV-1 and HIV-2, and also has activity on chronically infected cells and HIV strains resistant to reverse transcriptase inhibitors. Saquinavir is characterised by a low bioavailability and the metabolism is mainly hepatic through cytochrome P450. Resistance has been observed after both in vitro and in vivo drug exposure, with a relatively specific mutation profile compared with other protease inhibitors. Saquinavir is generally well tolerated, with mild gastrointestinal symptoms representing the most commonly observed adverse effects [21].
Ritonavir was protease inhibitors that have revolutionised HIV therapy. The extent of oral absorption is high and is not affected by food. The drug is primarily metabolised by cytochrome P450 isozymes and, to a lesser extent, by CYP2D6. Four major oxidative metabolites have been identified in humans, but are unlikely to contribute to the antiviral effect [22].
Indinavir is a synthetic peptidomimetic competitive inhibitor of the HIV aspartyl protease and their major limitation is the drug accumulation and no inductive effect on the hepatic CYP enzymes affecting its own metabolism [23]. In human urine, seven metabolites of indinavir have been detected and characterized [24]. The main adverse event of indinavir is nephrolithiasis, which is a result of the precipitation and crystallization of unmetabolized indinavir monohydrate in the renal tubules. Gastrointestinal complaints, as well as metabolic alterations, in particular hyperbilirubinemia, are among the other most common adverse events [23,25]. Significant data have demonstrated that indinavir has potent antiretroviral activity, although has pharmacologic characteristics that are unfavorable for chronic treatment and have been abandoned due to their significantly lower efficacy and greater toxicity than others PIs [26]. These obstacles have been overcome with the coadministration of low-dose ritonavir, which significantly improves its pharmacokinetic parameters [23].
Nelfinavir is a selective inhibitor of HIV protease, the enzyme responsible for post-translational processing of HIV propeptides. A unique mutation of the protease gene confers resistance. Diarrhoea is the most frequently reported adverse event in patients receiving nelfinavir-based combination therapy, together with rash, nausea, headache and asthenia Nelfinavir is metabolised by the cytochrome P450 system [27]. Although less effective than other PIs, nelfinavir is positioned as an alternative agent for the treatment of adults and adolescents with HIV infection and is an option for those unable to tolerate other protease inhibitors, as well as in pregnant and paediatric patients with HIV infection, due to prolonged viral suppression, good tolerability, a unique resistance profile [28,29].
Amprenavir is a sulfonamide that targets the protease enzymes, inhibiting post-transcripcional processing of HIV proteins [30]. The antiviral potency of this compounds have been demonstrated with various laboratory and clinical HIV strain and low toxicity in human cell line expressing T-cell markers has been reported [31].
The HIV PIs from first generations were combined with a backbone of nucleoside analogues, a rapid and durable complete suppression of HIV replication could be obtained for the first time in many patients. This virological effect was followed by immune recovery and, more importantly by a dramatic reduction in the incidence of opportunistic events [32]. However, the clinical utility of the first generation of PIs was limited by low bioavailability and high pill burdens, which ultimately reduced adherence and limited long-term viral inhibition [16].
2.2. Second-Generations of PIs
When therapy failure with first generation of PIs occur multiple protease resistance mutations were observed, often resulting in broad class resistance. To combat PI-resistance development, second-generation approaches have been developed in the early 2000s, including the lopinavir (Aluviran® and Koletra®) in 2000 and fosamprenavir (Lexiva® and Telzir®) and atazanavir (Reyataz®) in 2003 [16]. Its efficacy, tolerability and virological effect had been demonstrated.
Lopinavir is the most widely and lengthily testes PI. The drug was specifically designed to overcome the shortcomings of earlier agents in this class. The incidence of gastrointestinal disturbances is the main adverse effects together with lipid abnormalities [32]. Lopinavir is the only PI coformulated with ritonavir (Kaletra®), whose pharmacological boosting effect results in a highly potent, well-tolerated, clinically effective antiretroviral agent with a high genetic barrier to resistance [33].
Fosamprenavir is an inactive, highly water-soluble phosphate ester prodrug of amprenavir that contains a sulfonamide moiety [34]. This compound was synthesized to allow decreased pill burden, which contributes to improved patient compliace and it does not have any food restrictions [35]. Fosamprenavir is completely hydrolyzed to amprenavir in the gut endothelium, which is absorbed and metabolized [8]. The drug cause a low resistance and adverse effects compared with others PIs [36].
Atazanavir, an azapeptide, shows better metabolic preofile, fewer drug interaction and improved posology than prior PIs. This drug is rapidly absorbed and metabolized by the hepatic cytochrome P450 [37]. Atazanavir is generally well tolerated with low adverse events, which indirect bilirrubin elevation is the most frequent side effects reported [37,38].
2.3. Third-Generation PIs
Despite the availability of a growing number of potent antiretroviral agents, efforts to completely suppress viral replication in patients with HIV-1 infection have been developed. Recently, a third-generation tipranavir (Aptivus®) approved in 2005 and darunavir (Prezista®) in 2008 received full marketing authorisation, which present significantly greater potency and genetic barrier for resistance than all other PIs [32].
Tipranavir is a dihydropyrone and the unique non-peptidic nature HIV PIs that contains a sulphonamide moiety [8]. This compound remain active against most HIV strains resistance to others PIs [39] due to their structural flexibility, which allows it to fit into the active pocket of the protease enzyme in viruses that have become resistant to other PIs [40]. Tipranavir appears to have many drug-drug interactions and acts as an inhibitor of CYP isoenzymes [8].
The latest PI approved is the darunavir, an oral nonpeptidic HIV-1 PI that selectively inhibits the cleavage of HIV gag and gag-pol polyproteins, thereby preventing viral maturation. Althougth this compounds has some chemical similarities to amprenavir, it binds approximately 100-times more tightly to wild-type Darunavir is highly potent against laboratory strains and clinical isolates of wild-type and multidrug-resistant HIV and has limited cytotoxicity. The drugs did not display cross-resistance with other PIs and is primarily metabolized by the hepatic cytochrome P450 enzymes. The treatment has been generally well tolerated with most events being mild to moderate in severity, including diarrhoea, nausea, headache, upper respiratory tract infection and nasopharyngitis [41].
3. ASPARTIC PROTEASES: SOME PRELIMINARY EVIDENCES IN OPPORTUNISTIC PROTOZOAN PARASITES
The proteinases are involved in key areas of the relation between infectious agents and host; including antigen presentation, effectors cell function and tissue dissolution and remodelling, it is somewhat surprising that the proteinase inhibitors of parasite origin have not generally been the subject of intense research effort [42].
Aspartic proteases are a highly conserved family of proteins whose principal characteristic include a bilobal structure and the use of a catalytic dyad of aspartate residues (one in each lobe of the enzyme) to activate a nucleophilic water molecule and catalyze peptide cleavage [43]. Aspartic proteases are generally synthesized as zymogens and subsequently activated by proteolytic cleavage of the inhibitory proregion. They are found in eukaryotes and viruses, and have been attributed to a broad range of functions, from nutrient degradation to the activation of signaling molecules. A number of aspartic proteases have been implicated in virulence, for example, the HIV-.1 protease, the secreted Saps of Candida albicans or the hemoglobin degrading proteases of parasites ranging from P. falciparum to Schistosoma mansoni [44].
Aspartic proteases can be divided into a number of families. Most aspartic proteases described to date belong to family A1, for which the type member is pepsin; the other well-characterized aspartic protease families are the family A22 (type member presenilin) and viral family A2 (type member HIV-1 protease) [44]. For this last, several inhibitors have been designed to mimic endogenous peptides and thereby block the active site of aspartyl protease, a retroviral enzyme that cleaves the viral gag-pol polyprotein (Fig. 2). Consequently, the drugs prevent the HIV protease from carrying out its normal function, which is the proteolytic processing of precursor viral proteins into mature viral proteins [3]. Intrinsic antiviral potency (named as instantaneous inhibitory potential) could be greater for these PIs than for other antiretroviral drug classes [45]. The basis for this observation relies on the fact that nucleoside analogues and integrase inhibitors target reactions in which a single complex of enzyme and viral nucleic acid mediates a key step; by contrast, PIs target the whole number of enzymes themselves [32]. This action prevents the production of infectious viral particles with the subsequent decreased of the viral load and the partial immune restoration [46].
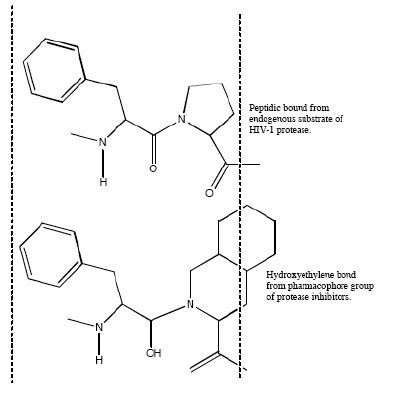
Mimetic action of hydroxyethylene bond from active group of peptidic protease inhibitor with normal peptide linkage cleaved by the human immunodeficiency virus protease.
Protease enzymes seem to play important roles in the life cycles of all medically important protozoan parasites and are utilized for diverse functions, including the invasion of host cells and tissues, the degradation of mediators of the immune response and the hydrolysis of host proteins for nutritional purposes. Between them, the aspartic proteases play an important role in physiological and pathology process caused by protozoa. Their biochemical and molecular characterization have been providing tools to improve our understanding of the functions of these enzymes. In addition, studies in multiple systems suggest that inhibitors of protozoan proteases have potent antiparasitic effects, which have been demonstrated by some clinically useful drugs [42,47,48].
Within the apicomplexa, has been proposed that there are two different families of aspartic proteases with distinct evolutionary lineages. The first, more ancient group includes certain aspartic proteases of Eimeria tenella (eimepsin), and P. falciparum (plasmepsin VI). The second group may have been acquired by horizontal transfer and is composed of the hemoglobin-degrading enzymes of Plasmodium spp. such as plasmepsins I, II, and IV [49]. However a recent study identified five main groups of apicomplexan aspartic proteases using phylogenetic analysis, the best tool which allows the classification of the different proteins into evolutionary related families, that frequently also have related biological functions [44].
For P. falciparum, 10 putative aspartic proteases have been described (PfPMI to PfPMX) [50]. The best characterized of these are the hemoglobin degradading plasmepsins: PfPMI, PfPMIV and histoaspartic protease (HAP). These four proteases are expressed during the intraerythrocytic life cycle stages and have been localized to the acidic food vacuole, where they are responsible for the first steps in hemoglobin degradation [51].
For T. gondii, seven genes coding for putative aspartic proteases, identified in the complete genome by homology with know human and apicomplexan proteases. TgASP1, TgASP3 and TgASP5 genes have been experimentally annoted by cDNA sequencing; the other TgASPs were assembled using expressed sequence tag databases and manual annotation based on homology [44].
Although there are no report of aspartic proteases in C. parvum, Mele et al. 2003 [52] identified several different C. parvum sequences that showed homology with this protein family when the eimepsin sequence was used as an electronic probe on the C. parvum genomic database. Additionally, Morales Gomez (2004) [53] also identified several different C. parvum sequences that showed homology with a protein family related to aspartyl proteases.
Additional studies in parasites, to identify all the possible sequences related with proteases will be needed. The possible structural relation, similarity in sequences or function between these enzymes with the HIV-1 aspartic protease could explain the effect of HIV-1 PIs against some opportunistic protozoan, even though these last, were formulated with unique specificity for HIV-1 aspartic protease.
4. HIV-1 PIS AS ANTIPROTOZOAN AGENTS
The drastic reduction in the incidence, morbidity and mortality of AIDS coinfections after the introduction of PIs in the antiretroviral therapy was the first line of evidence that these compounds could exert a direct effect on opportunistic pathogens [54]. Although there are several studies concerning protozoa parasites and HIV co-infection that including particularities of its epidemiology, pathogenesis, prophylaxis, the direct incidence of treatment on parasite burden remain unclear and undefined. In this sense, in vitro and in vivo studies have been conducted to recognize the effects of HIV-1 PIs against protozoan opportunistic parasites. Even though, the studies are scarce, the preliminary evidences suggest the unspecific activity of these PIs on some parasites.
In this article, we review the reports about the antiprotozoal activity of PIs. Data was selected from medical literature published in any language using Medline since 1995 to 2009 for published literature. Search terms were each PI combined with all protozoa parasite with medical importance. The search strategy yielded a total of 27 articles during the last 15 years, including one review article. After initial screening, nine articles were excluded which did not evaluated antiprotozoal activity of PIs (8) or did not provided the necessary data (1). The data recovered by year of publication shown an increase of articles published in the last three years (Fig. 3).
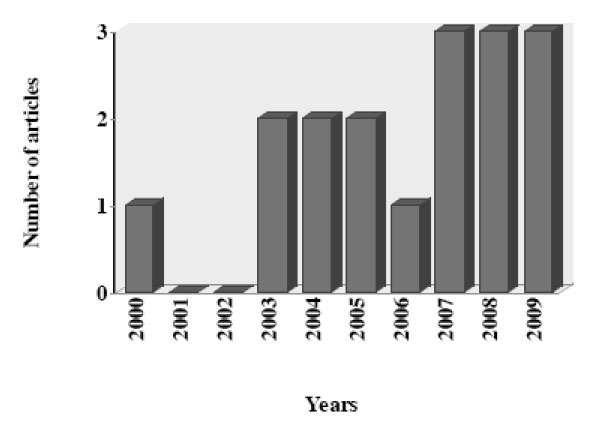
Number of articles about antiprotozoal activity of HIV-protease inhibitors by year (N = 26).
The analyses indicated that the PIs studied have been saquinavir, ritonavir, indinavir, nelfinavir, lopinavir and atazanavir; while no reports were found with amprenavir, fosamprenavir, tipranavir and darunavir. The genus of opportunistic protozoa involved in the studies have been Plasmodium, Leishmania, Toxoplasma, Trichomonas, Giardia, Cryptosporidium and Microsporidium, which are in correlation with the protozoa that cause major morbidity in HIV patients (Table 1).
Studies by HIV-Protease Inhibitor in Relation to their Antiprotozoal Action (01/1995 – 12/2009)
| Protozoos | Saquinavir | Ritonavir | Indinavir | Nelfinavir | Lopinavir | Atazanavir |
|---|---|---|---|---|---|---|
| Plasmodium spp. | 8 | 9 | 1 | 2 | 5 | 1 |
| Leishmania spp. | 2 | 1 | 1 | 2 | 1 | 0 |
| Toxoplasma gondii | 0 | 1 | 0 | 1 | 0 | 0 |
| Trichomonas vaginalis | 1 | 1 | 0 | 0 | 1 | 0 |
| Giardia lamblia | 1 | 1 | 0 | 0 | 1 | 0 |
| Cryptosporidium parvum | 2 | 2 | 3 | 1 | 0 | 0 |
4.1. Activity Against Plasmodium spp
The World Health Organization estimates that 300-500 million cases of malaria and 1-2 million deaths due to malaria occur annually [55]. When HIV and malaria parasite are present as a coinfection, they enhance each other pathogenicity [56,57]. At the clinical level, the interaction between HIV and malarial infection is complex and bidirectional [58]. Although the morbidity attributable to each infection in the presence of the other is not fully understood, interactions that are likely to have significant clinical consequences have been described [59, 60]. Interestingly, lines of evidences suggest a possible beneficial action of some antiretroviral PIs, commonly used in HAART, as antimalarial agents.
In this sense, two HIV-1 PIs have been reported to reduce in vitro cytoadherence of P. falciparum-infected erythrocytes to drug-exposed endothelial cells [61], a process linked in vivo with parasite virulence. More recently, Skimer–Adams et al, 2004 [62] indicated that three PIs (saquinavir, ritonavir and indinavir) directly inhibited the grown of P. falciparumin vitro at clinically relevant concentrations, suggesting that, some PIs are active against the most virulent human malaria parasite, which express a number of aspartic proteases; know as plasmepsins [51]. This finding was corroborated by Parikh et al. (2005) [63] when tested the activity of seven HIV-1 PIs against P. falciparum, evidencing that all compounds inhibited the development of parasites at pharmacologically relevant concentrations.
Combinations of these antiretroviral PIs in a murine model of malaria have been also examined, showing a delay in patency and a significant attenuation of parasitemia in mice infected with Plasmodium chabaudi and treated orally with ritonavir-saquinavir or ritonavir-lopinavir. In addition, in vitro enzyme assays demonstrated that P. falciparum plasmepsins II and IV are both inhibited by the antiretroviral PIs saquinavir, ritonavir and lopinavir [64].
On the other hand, the synergist between HIV-1 PIs and antimalarial drugs had been also investigated. A previous study suggested that saquinavir and ritonavir behaved synergistically with chloroquine (CQ) against chloroquine-resistant line in vitro [65]. One year later, He et al. (2008) [66], found that all HIV PIs tested were able to enhance the antimalarial action of CQ. In both, in vitro and in vivo assays systems performed, ritonavir was found to be the most potent in patenting the antimalarial action of CQ. More recently, the mechanism by which the antiretroviral PIs modulate the CQ resistance in malaria parasites was investigated. The authors found that CQ-resistant parasites showed increased intracellular glutathione levels in comparison with the CQ-sensitive parasites. Treatment with some antiretroviral PIs significantly reduced the levels of glutathione (GSH) and glutathione S-transferase activities in CQ-resistant parasites. Ritonavir also decreased glutathione reductase activities and glutathione peroxidase activities in CQ-resistant parasite line. At respect the authors suggest that HIV PIs (ritonavir, saquinavir, nelfinavir), especially ritonavir, are able to enhance the sensitivity of malaria parasites to CQ by suppression of GSH levels and alteration of the related enzyme activities [67].
Finally, a very recent study indicated that the HIV-PIs lopinavir and saquinavir affect preerythrocytic-stage parasite development in vitro. When the effects of HIV-PIs on preerythrocytic-stages in vivo were investigated the results showed that lopinavir/ritonavir had a dose-dependent effect on liver-stage parasite development [68].
Through, the mechanism responsible for the antimalarial activity of these antiretroviral PIs is not fully clear, the preliminary data suggesting that select protease inhibitors will offer some protection against malaria directly, or potentiating the antimalarial action of drugs commonly used but with reduced efficacy due to the resistance.
4.2. Activity against Leishmania spp
Leishmaniasis is among the most neglected of the tropical diseases: more than 12 million people are currently infected worldwide, there are 2 million new cases every year and 350 million people are considered to be at risk [69].
Two basic clinical forms are recognized –cutaneous leishmaniasis (CL), a disfiguring and stigmatizing disease, and visceral leishmaniasis (VL) or kala azar, which is fatal without treatment [69]. Current therapy is suboptimal due to toxicity of the available therapeutic agents and drug resistance [70]. In addition, the emergence of AIDS pandemic and the occurrence of HIV-Leishmania co-infection are added as a new complex problem.
Although there are several studies concerning Leishmania/HIV co-infection, some particularities of its epidemiology, pathogenesis, prophylaxis and especially of its treatment remain unclear and undefined [69]. With the introduction of PIs in the antiretroviral therapy for HIV-infected individuals, the number of co-infected cases reported in European Leishmania-endemic countries fell sharply. Nevertheless, the disease is still the third most frequent parasitic opportunistic infection in Europe [71].
As far as we know, only three articles have been evaluated the antileishmanial activity of HIV-1 PIs. In 2005, two antiretroviral PIs (indinavir and saquinavir), described a dose-dependent leishmanicidal activity in vitro, which was greater on L. major than on L. infantum promastigotes [72]. The authors reported that the inhibition of leishmanial growth produced by these compounds may be due to their effects on proteasome; which constitute a large, non-lysosomal, multi-subunit protease complex presents in Leishmania, related with the differentiation and replication of protozoan [73].
Three years later, Trudei et al. (2008) [74] analyzed the efficacy of 3 HIV PIs (nelfinavir, ritonavir and saquinavir) in controlling the intracellular growth of Leishmania parasites in the human monocytic cell line THP-1 and, more importantly, in human primary monocyte-derived macrophages (MDMs). The results showed that intracellular survival of both L. infantum and L. donovani markedly reduced after treatment with PIs. Interestingly, antileishmanial activity of PIs was still seen even when infection was done with a Leishmania field isolate that is resistant to sodium stibogluconate (SbV), the drug most commonly used for the treatment of leishmaniasis [74].
Finally, very recently, a very complete study analyzing the effect of aspartyl peptidase inhibitors on the L amazonensis proliferation, ultrastructure, interaction with macrophages cells and expression of classical peptidases which are directly involved in the Leishmania pathogenesis was performed.
The results showed that all HIV-1 PIs (lopinavir, nelfinavir, amprenavir, indinavir, saquinavir) impaired parasite growth in a dose-dependent fashion, especially nelfinavir and lopinavir [70]. The authors pointed that the inhibitory effect of HIV-1 PIs in vitro were observed at substantial PIs concentrations (µM range), much higher than those needed for HIV peptidase inhibition (on the nM order), probably reflecting a much lower affinity of these drugs for a yet unidentified target in Leishmania compared with their affinity for HIV peptidase [15].
Interestingly, the research evidenced that HIV-1 PIs treatment caused profound changes in the Leishmania ultrastructure, such as cytoplasm shrinking, a higher number of vesicles, nucleus surrounded by endoplasmic reticulum, mitochondrial swelling and myelin-like structures, blocks of condensed chromatin [70]. These changes were dependent of the PIs (nelfinavir or lopinavir) used or its combination. In addition, the hydrolysis of HIV peptidase substrate by L. amazonensis extract was inhibited by pepstatin and HIV PIs, suggesting that an aspartyl peptidase may be the intracellular target of the inhibitors. Nevertheless, despite these beneficial effects, the HIV PIs induced an increase in the expression of cysteine peptidase b (cpb) and the metallopeptidase gp63 [70], two well-known virulence factors expressed by Leishmania spp [75,76]. At respect future studies should be needed to understand and to define if the use of HIV-1 PIs could be beneficial or not in the treatment of leishmaniasis.
4.3. Activity Against T. gondii
T. gondii is a widely distributed apicomplexan parasite of great medical importance. Most primary infections are asymptomatic, and may exist in up to one-third of the human population worldwide [77]. In immunocompromised patients, especially those with AIDS, reactivation of latent infection causes necrotic lesions producing life threatening encephalitis [78]. The majority of these cases are thought to result from recurrence of chronic infections, due to the decrease in cellular immunity [79].
Until present, Toxoplasmic encephalitis (TE) still represents the most common cerebral mass lesion in patients infected with HIV. However, of the five longitudinal studies that have estimated the occurrence of TE, both before HAART and since its introduction, four have reported that the use of HAART in which PIs were included has resulted in marked decreases in TE among HIV positive individuals [54].
Preliminary evidences showing direct pharmacological effect on T. gondii growth using ritonavir and nelfinavir [7] were reported. These PIs were highly inhibitory for Toxoplasma growth, with concentrations that can be archived in humans [15]. Interestingly, none of the PIs tested in this research affected the anti-Toxoplasma activity of pyrimethamine or sulfadiazine in vitro, the first line of anti-Toxoplasma drugs.
The mode of action of HIV-1 PIs on T. gondii remains to be elucidated and future studied defining the mechanism involved in the inhibition of T. gondii growth by PIs could allow define the specificity of HIV-1 PIs against another protozoan parasites proteases.
4.4. Activity Against Giardia and Trichomonas vaginalis
Trichomonas vaginalis is an important protozoan parasite transmitted principally through vaginal intercourse. Despite a relative paucity of studies of the prevalence and incidence of trichomoniasis, recent publications suggest that T. vaginalis is one of the most common sexually transmitted infections worldwide with over 170 million cases per year [80,81]. It is now recognized that Trichomonas may play a critical and underrecognized role in amplifying HIV transmission, and it may have a major impact on the epidemic dynamics of HIV in African-American communities.
In the context of giardiasis, the acute stage, posterior to infection with this parasite can last from 3 to 4 days, but can persist for much longer. Persistent infection and diarrhea may occur in immunocompromised individuals, for example IgA deficiency, but is less obvious in cases of HIV individuals [82]
Metronidazole is the drug of choice to treat both medically important parasites although resistance, particularly in T. vaginalis is of great concern [83]. For giardiasis, even though, there are alternatives to metronidazole, treatment failures with all commonly used drugs are well documented [84].
The use of PIs could allow a better clinical management in T. vaginalis or Giardia spp infections. Nevertheless, the possible utility of these inhibitors is practically unexplored. As far as we know; only one study had been investigated the in vitro activity of Kaletra (co-formulation of lopinavir and ritonavir), ritonavir and saquinavir against G. duodenalis and T. vaginalis. Kaletra was the most effective overall, with 50% effective. Within the range of human plasma concentration for ritonavir, only G. duodenalis was inhibited. Lopinavir alone was less inhibitory than ritonavir but was associated with a blockage in cytoquinesis of G. duodenalis trophozoites. Additionally, saquinavir was not effective against these protozoans [85].
Further investigations, confirming beneficial effects of HIV-1 PIs against T. vaginalis and G. duodenalis are essentials. The elucidation of possible targets for the HIV-PIs in these parasites could be essential to develop new drugs to protect against infection by both parasites.
4.5. Activity Against Cryptosporidium, Microsporidium and Isospora Belli
Infection with the apicomplexan parasite Cryptosporidium parvum leads to self-limiting diarrhoea in immunocompetent individual, but can develop into a severe and chronic disease in immunocompromised patients [86]. With the introduction of PIs as part of the HAART in the treatment of HIV infections, a beneficial effect was noted in AIDS patients suffering from cryptosporidiosis [87,88]. At respect it has been hypothesised that protease inhibitors (PI) could play a chemotherapeutic role in controlling cryptosporidiosis, given that protease activity has been observed during several of the developmental stages of C. parvum and that the proteases of C. parvum are believed to be essential for the host cells to become infected [89-92]. However, direct antiparasitic effects of antiretroviral PIs on C. parvum have not been sufficiently explored, as for the most of protozoan opportunistic parasites. Even though it has been concluded that the effect of these antiretroviral PIs on infections with C. parvum is secondary following immunological restoration [87] and none aspartyl protease have been described functionally in C. parvum several different C. parvum sequences that showed homology with a protein family related to aspartyl proteases were previously described [52,53]. Future studies could to link these proteins as possible targets for HIV PIs.
Preliminarily a recent study showing that indinavir directly interferes with the cycle of C. parvum, was reported and a marked reduction in oocyst shedding and in the number of intracellular parasites was observed [52]. In the same year, the effect of five PIs used in HAART on the excystation, invasion and development of this parasite in a cell culture system were tested. The results showed that indinavir, nelfinavir and ritonavir inhibited parasite development significantly. The inhibitory effect was increased when the aminoglycoside paromomycin was combined with the PIs indinavir, ritonavir, and to a lesser extent saquinavir, compared to the PIs alone [86].
More recently, Human ileocecal adenocarcinoma tumor cells (HCT-8) were used as in vitro model to evaluate the activity of indinavir on C. parvum. The results demonstrated that PIs directly exert an inhibitory effect on this parasite and the extent of this effect depended on the specific dose and the duration of treatment. In addition several different C. parvum sequences that showed homology with a protein family related to aspartyl proteases were identified [53]. At respect, the authors pointed that although there are no reports of aspartyl proteases in C. parvum, the inhibitory effect of PIs on C. parvum growth in vitro suggests that aspartyl proteases could have some important functions for this parasite. In fact, proteolytic activities have been demonstrated during peak periods of excystation in C. parvum oocysts and cysteine and serine protease classes have been functionally associated with this process [53].
For Microsporidium and Isospora belli, until now, none assay detecting the possible effect of HIV-1 PIs on this parasite has been performed. The identification of in vitro and in vivo activities of these inhibitors, in combination with the study of the aspartic proteases enzymes for both parasites could contribute to find clinical and microbiological interesting findings.
5. CONCLUSIONS
The introduction of the PIs as part of the HAART in the treatment of HIV-infections has been evidenced an impact in the AIDS disease evolution and the outcome of opportunistic infections. Although, other formulations are included in the HAART, and the final effect of the treatment is based on the combination of all the inhibitors (proteases and transcriptase inverse), the possible effects of PIs against other opportunistic pathogens, indicate their efficacy and multiplicity of functions. The HIV antiretroviral PIs were developed with unique specificity for the HIV-1 aspartyl protease, but could be possible their specificity for another parasitic proteases. In addition, even though this possibility could be reasonable, unrelated effects of HIV PIs on some protozoan should be considered. Additional studies should be needed to provide valuable information on ultrastructural modifications and toxic effect on the cells exposed to these PIs. The sustrate –inhibitors specificity studies are crucial to define the specific mechanism for the inhibition of other proteases, even though these PIs were formulated with unique specificity.

