All published articles of this journal are available on ScienceDirect.

Development of a Peptide-Derived Orally-Active Kappa-Opioid Receptor Agonist Targeting Peripheral Pain
Authors Info & Affiliations
Abstract
Kappa-opioid agonists are particularly efficacious in the treatment of peripheral pain but suffer from central nervous system (CNS)-mediated effects that limit their development. One promising kappa-agonist is the peptidic compound CR665. Although not orally available, CR665 given i.v. exhibits high peripheral to CNS selectivity and benefits patients with visceral and neuropathic pain. In this study we have generated a series of derivatives of CR665 and screened them for oral activity in the acetic acid-induced rat writhing assay for peripheral pain. Five compounds were further screened for specificity of activation of kappa receptors as well as agonism and antagonism at mu and delta receptors, which can lead to off-target effects. All active derivatives engaged the kappa receptor with EC50s in the low nM range while agonist selectivity for kappa over mu or delta was >11,000-200,000-fold. No antagonist activity was detected. One compound was chosen for further analysis (Compound 9). An oral dose response of 9 in rats yielded an EC50 of 4.7 mg/kg, approaching a druggable level for an oral analgesic. To assess the peripheral selectivity of this compound an i.v. dose response in rats was assessed in the writhing assay and hotplate assay (an assay of CNS-mediated pain). The EC50 in the writhing assay was 0.032 mg/kg while no activity was detectable in the hotplate assay at doses as high as 30 mg/kg, indicating a peripheral selectivity of >900-fold. We propose that compound 9 is a candidate for development as an orally-available peripherally-restricted kappa agonist.
INTRODUCTION
Pain is the most common symptom that leads people to seek medical intervention in the United States [1]. While estimates of people with chronic pain vary widely, a 2001 study revealed that 50% of individuals reported having ‘any’ chronic pain in the preceding three months, 14% reported having ‘significant’ chronic pain while 6% reported having ‘severe’ chronic pain [2]. These values increase with age, cancer and in patients that are hospitalized. The great majority of general practitioners (81%) believe that pain management is ineffective in over half of the patients seeking help [2]. The most difficult pain to manage successfully is chronic pain, which includes visceral, thermal, bone, and neuropathic pain, and pain associated with cancer [3]. Currently, there are two major types of chronic pain medications in use – opioids and non-opioids – both of which have important limitations. Non-opioid analgesics include paracetamol and the NSAIDs, all of which target prostaglandin formation, usually through the inhibition of the COX-1 and COX-2 enzymes. Nonselective COX inhibitors result in adverse side-effects associated with COX-1 inhibition, including renal dysfunction, GI ulceration and inhibition of platelet aggregation. Selective COX-2 inhibitors were developed fairly recently, but have been associated with increased thromboembolytic risk which has decisively limited their use [4]. These compounds target inflammation-associated pain which may or may not contribute to chronic pain states. Opioids are the main class of analgesics used in the treatment of moderate to severe chronic pain. These compounds have various side effects including nausea, vomiting, constipation, depressed breathing and neurotoxicity. Most significantly, patients can become both addicted and tolerant to these agents, thus requiring dose escalations to maintain therapeutic value.
The mediation of opioid analgesic effects occurs through three receptors, mu-, kappa-, and delta-. Activation of these receptors was long thought to occur only centrally, but in recent years, the receptors have been found in peripheral sensory neurons that can be modulated by endogenous opioids or opioid drugs [5]. Opioids also have anti-inflammatory properties, hence they exhibit more pronounced analgesic effects in damaged (inflammatory) versus normal tissues. This appears to be a result of upregulation of the opioid receptors during inflammatory events and release of opioid peptides (endorphins, enkephalins, dynorphins and others) from immune calls. Mechanical nerve damage leading to neuropathic pain also results in the upregulation of the opioid receptors, and corresponding greater opioid analgesic effects. Endogenous opioid peptides are released in response to damage of stimulatory neurons and by immune cells in response to inflammation, which is consistent with a regulated response to inflammation and attendant pain. Finally, recent studies suggest that systemically and centrally administered opioids may be acting predominantly (50-80%) as agonists of the peripheral opioid receptors [6,7].
Opioids differentially target the three opioid receptors, both in the CNS and peripherally, which can lead to untoward side-effects. Agonists at the mu-receptor are the most currently used opioids, but suffer from induction of euphoria, addiction, respiratory depression, and GI tract inhibition. Kappa opioid agonists (KOAs) exhibit none of these effects, and have been shown in visceral pain models to be the most efficacious of the opioids [6,8]. However, evaluation of early KOAs, including non-peptidic compounds U50,488, enadoline, ADL 10-0101 and ADL 10-0116, demonstrated a poor peripheral versus central activity that resulted in centrally-mediated sedation and dysphoria. These side effects resulted in discontinuation of their development as analgesics [6]. Potentially more promising are peptidic compounds, including FE20041 and FE200665, which exhibit very high peripheral versus central activity and have shown benefit in patients with visceral and neuropathic pain, having the same analgesic effects as the early KOAs without the negative side-effects [8-12].
FE200665 (renamed CR665) has been under evaluation as a peripherally selective KOA for pain by Cara Therapeutics (Shelton, CT) [7,13]. This compound is not orally active, which drastically restricts its potential use as a broad-spectrum analgesic for peripheral pain. Our laboratory has developed a number of proprietary modifications to the cationic amino acids arginine (Arg) and lysine (Lys) that impart stability and oral availability when incorporated into peptides [14]. In this study we have synthesized a series of analogs of CR665 encompassing modifications to the D-Arg that is residue 4 in the compound. These derivatives were screened for efficacy and oral availability using the rodent acetic acid-induced writhing model that evaluates peripheral pain modulation, the model employed by Cara Therapeutics. Five compounds were further screened for specificity of activation of kappa receptors as well as agonism at mu and delta receptors. Finally, one compound was assessed for oral dose response in the writhing assay as well as i.v. dose response in writhing and hotplate assays (which evaluate peripheral versus CNS-derived pain). This compound was found to be highly specific for kappa receptors with little or no agonist or antagonist activity at mu or delta receptors, has a druggable oral dose response, and a >900-fold peripheral selectivity.
MATERIALS AND METHODS
Synthesis of Analogs
The Position 4 D-Arg residue of CR665 was converted to derivatives containing the modified D-Arg or D-Lys residues shown in (Fig. 1). The strategy was to slightly modify the structure of the proven active compound CR665 to see if improvements in oral activity and selectivity against peripheral pain can be elicited. This strategy has been applied successfully in our laboratory, resulting in the identification of lead compounds for psychosis, stroke and neuropathic pain in which their pharmacokinetic/pharmacodynamic (PK/PD) properties were drastically improved[14-16]. Compounds 1-4 feature alkylations of the D-Arg residue whereas compounds 5-18 feature modifications to the side-chain length and extent of alkylation of a hypothetical Position 4 D-Lys residue (which was not evaluated previously [7]). For the D-Lys compounds, we have found in earlier studies that a Lys derivative often improves PK/PD when substituted for an Arg residue in the parent compound [14-16]. All compounds were assembled using standard Merrifield chemistry to build the peptides while incorporating the non-natural D-Arg and D-Lys residues, synthesized as described previously [17-22]. All compounds were HPLC-purified and characterized by MALDI-mass spectrometry, giving measured [M+1]+ molecular weights within 0.1% of theoretical values. This is the standard method of analysis for peptides synthesized through standard peptide synthetic routes, in which the identity of each monomer is certain.
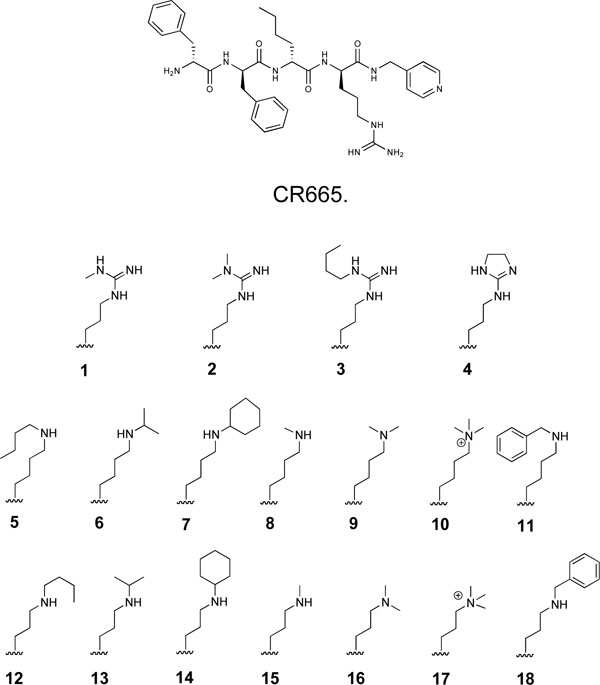
Structure of CR665 and side-chain structures of proprietary CR665 derivatives.
Animals
All animal work was reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) at the Medical University of South Carolina. All experimental protocols were performed in accordance with the guidelines set forth in the NIH Guide for the Care and Use of Laboratory Animals, published by the Public Health Service. All protocols were performed on male Sprague-Dawley rats (Harlan, Prattville AL, 240-280g), which were housed in an AAALAC-approved colony room maintained at a constant temperature and humidity. Animals (two per cage; approximately 175-225 g each) were kept on a 12-h light:dark cycle with ad libitum access to food and water. Because many of the assays are susceptible to learning phenomena, which results in progressive decrease of the physiological response or behavior to be monitored, rats were used no more than three times per analgesic assay.
Pain Models
Acetic Acid-Induced Writhing Model
This protocol is a well-established model of peripheral pain [11,23,24]. Following oral or i.v. administration of compound, rats are rested for 20 min before i.p. injection with 2 ml/kg of 3% acetic acid. Animals are then rested an additional 10 min before being placed in a 10 x 10 inch chamber. A video of the rats is then recorded for next 20 min. The video is subsequently blindly scored by an investigator who examines each rat every 20 sec for the entire 20 min (60 individual observations). At each observation rats are scored on whether they are writhing or not. Writhing is defined as a constriction of the abdominal area, often with extension of the hind legs. At the end of the experiment the percent of the time the animal was writhing was calculated. The dose-response curves and EC50 values were generated using GraphPad Prism®.
Hotplate Model
The hotplate model evaluates central pain attenuation in a rodent after applying an acute thermal stimuli [25-27]. The rat is treated with compound and assessed over time on a hotplate analgesia meter (Columbus Instruments, Columbus, OH), essentially a flat surface maintained at 53.0+/-0.2°C. The time until the rat lifts, nibbles or shakes one of its hind paws is recorded, which is known as the response latency. Animals remain on the hotplate no longer than 30 sec to avoid tissue damage. Experiments are scored as the percent of maximal possible effect (%MPE) calculated using the following equation: %MPE = [(post-drug latency – pre-drug latency)/(cut-off pre-drug latency)] x 100%. The dose-response curves and EC50 values were generated using GraphPad Prism®.
Opioid Receptor Activation Assay
PathHunter™ Beta-Arrestin GPCR Functional Profiling and Screening cell lines that express the individual opioid receptors were obtained from DiscoveRx (Fremont, CA). These cells express the kappa (or mu or delta) opioid receptor fused to a proprietary β-galactosidase enzyme fragment and the β-arrestin gene fused to an enzyme acceptor (the remaining fragment of β-galactosidase). Upon activation of the receptor, β-arrestin is recruited to the GPCR and the β-galactosidase is activated through enzyme complementation. Cells were grown using standard cell culture techniques and then plated at 30,000 cells/well in 90 µl in a 96 well white, clear bottom plate and incubated overnight. Dilutions were prepared in dilution buffer (HBBS, 20 mMHepes, 0.1%BSA) and 10 µl added per well. Plates were then incubated for 90 min at 37°C in a water-saturated atmosphere. Plates were then allowed to come to room temp and DiscoveRx ‘s proprietary developing agent added as recommended by the manufacturer. This agent contains a detergent, 6-O-β-galactopyranosyl-luciferen and luciferase. Plates were then incubated for 1 hr at room temperature before being read on a luminometer. Antagonist studies were performed in an identical manner except after the overnight incubation 10 µl media was removed and replaced with drug or naloxone at the indicated concentration. Cells were then incubated for 30 min at 37°C in a water-saturated atmosphere before addition of 10 µl of the indicated agonist (lys-dermorphin for mu receptors and [D-Ala2, D-Leu5]-enkephalin (DADLE) for delta receptors).
All plates had wells treated with dilution buffer only (background) and others treated with a concentration of agonist shown in preliminary studies to be maximal (200 nM; maximal luminescence). To calculate % maximal luminescence, background was subtracted from the experimental values and this was divided by maximal luminescence and multiplied by 100.
Statistics
To test for significant differences in the writing assay during the screening of doses (Fig. 2) a Student’s t-test was used comparing each compound individually to saline control. Results were considered significantly different if p<0.05. For all dose response experiments (opioid receptor binding assays and writhing dose response) EC50s and 95% confidence limits were calculated using Graphpad Prism software.
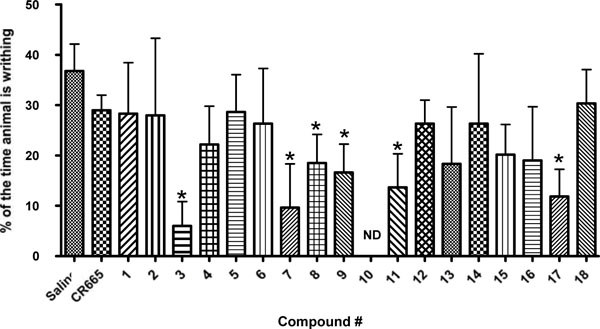
Screen of compounds 1-18 in the acetic acid-induced writhing assay in rats. Results are shown as the mean ± SEM with n = 3-9 for each compound. Asterisk indicates that the value is significantly different from the saline control value by Student’s T-Test, p<0.05.
RESULTS
Fig. (1) illustrates the series of KOA peptide derivatives that were synthesized and evaluated. All feature modifications to the Position 4 D-Arg residue, and were chosen to provide a range of structures to initially probe potential structure-activity relationships (SARs) at this position. Compounds 1-4 are modified D-Args, compounds 5-11 are modified D-lys while compounds 12-18 are D-Orns (or 5-11 truncated by one methylene group in the side-chain).
Screening of Potential Orally-Available KOAs
As an initial biological screen the potential for oral availability of the 18 peptides in our library were evaluated in a well-established model of peripheral pain, the acetic acid-induced writhing assay. Rats were administered the various compounds by oral gavage at a screening dose of 20 mg/kg 20 min before i.p. injection with 2 ml/kg of 3% acetic acid. 10 min later animals were assessed for writhing as indicated in the methods section. As shown in (Fig. 2), several of the compounds (3, 7, 8, 9, 11 and 17) were able to significantly block outward physical signs of peripheral pain in this assay (writhing) while the parent compound, CR665, did not have a significant effect. Thus, several of the Arg modifications can impart CR665 with the ability to cross the gut barrier. Compounds 3, 7, 9, 11 and 17 were selected for further analysis.
Analysis of Kappa Receptor Activation by the KOA Compounds
The current compounds, like CR665, are intended to specifically activate the kappa opioid receptor. To confirm this, and to determine their relative activities, we assessed their efficiency using PathHunter™ Beta-Arrestin GPCR Functional Profiling and Screening cell lines from DiscoveRx (Fremont, CA) as described in the Materials and Methods section. As shown in (Fig. 3), compounds 3, 7, 9, 11 and 17 activate the kappa opioid receptor with EC50s in the low nM range, as did CR665 and dynorphin (the positive control).
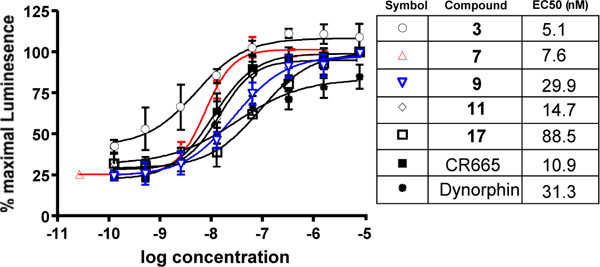
Dose response (and EC50) activation of kappa receptors by compounds 3, 7, 9, 11 and 17 in the DiscoveRx PathHunter™ Beta-Arrestin platform (95% confidence levels (nM); 3:1.38 to 18.67, 7:5.63 to 13.14, 9:15.34 to 54.48, 11:8.15 to 26.77, 17:47.2 to 166.0; CR665:9.07 to 13.14; Dynorphin:10.14 to 96.62). Individual points are the mean ± SEM of 3-6 separate repeats.
For a peripheral kappa agonist to be effective with minimal side effects it is important that it not interact to any substantial degree with other opioid receptors. Thus, we have used the cloned human receptors in the DiscoveRx system to assess activation at mu or delta receptors. As shown in (Fig. 4), none of our compounds were able to activate the mu (Fig. 4A) or delta (4B) opioid receptors, even at mM concentrations. This indicates a kappa specificity for these compounds from >11,000 fold for 17 to >33,400 for 9 to >200,000 fold for 3. These are minimal estimates; since there were no detectable mu or delta activity, the actual selectivities may be much higher. Lys-dermorphn and DADLE are included as positive controls and have EC50s in the nM range.
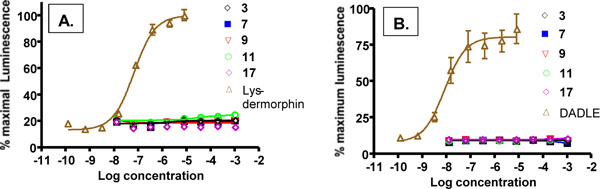
Activation of mu (A) and delta (B) opioid receptors by compounds 3, 7, 9, 11 and 17 in DiscoveRx PathHunter™ Beta-Arrestin platform. Lys-dermorphin and DADLE at 200 nM served as positive controls for mu and delta receptors, respectively. Individual points are the mean ± SEM of 3-6 separate repeats. No activation was detected.
To insure the compounds did not act as antagonists at the non-kappa opioid receptors, cells of the DiscoveRx system that express mu receptors exclusively were first incubated with compound (or the non-selective opioid antagonist naloxone as a positive control) followed by agonist (Lys-dermorphin or DADLE at 200 nM). As shown in (Fig. 5A), only slight antagonist effects on mu receptors were detected and then only at the 1 mM concentration. Likewise, similar studies were performed using DiscoveRx cells that exclusively express delta receptors. None of the tested compounds exerted any significant antagonism with these receptors (Fig. 5B). The effects of the naloxone in these studies were similar to published studies [28,29].

Antagonism of mu (A) and delta (B) opioid receptors by 3, 7, 9, 11 and 17 in the DiscoveRx PathHunter™ Beta-Arrestin platform. Naloxone served as a positive control antagonist for both mu and delta receptors whereas lys-dermorphin and DADLE at 200 nM were the control agonists, respectively. The mean ± SEM of cells given no additions of agonist and/or antagonists is indicated by the x and error bars next to the words “No add”. Individual points are the mean ± SEM of 3-6 separate repeats.
Oral Dose Response and Peripheral Selectivity of Compound 9
Five compounds, 3, 7, 9, 11 and 17 exhibited extensive yet statistically indistinguishable activity in the various in vitro and in vivo experimental evaluations (Figs. 2-6). Compound 9 was chosen for proof of concept studies to evaluate potential oral efficacy and peripheral selectivity, based on the ease of synthesis of its particular non-natural Lys residue compared to the other candidates [17,18]. As shown in (Fig. 6), the analgesic effect of compound 9 in the writhing model of peripheral pain when administered orally is dose-responsive with an EC50 of 4.7 mg/kg.
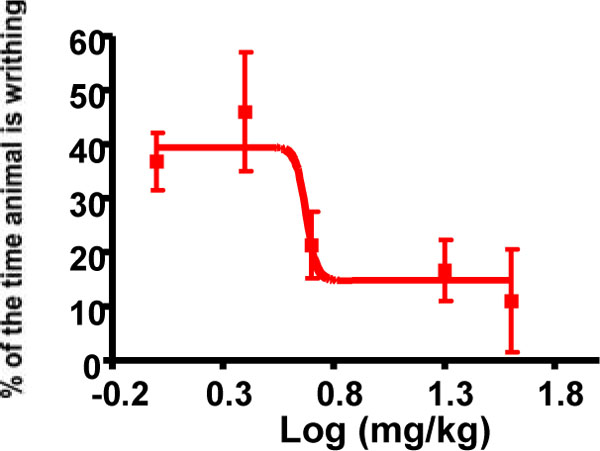
Oral dose-response evaluation of compound 9 in the acetic acid writhing model (EC50 = 4.7 mg/kg). Individual points are the mean ± SEM, n=3 for all data points.
As discussed, central availability of a kappa agonist can lead to undesirable side effects. To assess the peripheral to central selectivity of compound 9 an i.v. dose response of this drug was performed in both a model of peripheral pain (A. the writhing assay) and central pain (B. the hotplate assay). The ratio of EC50s for each of these assays can then be used to determine the peripheral selectivity of the drug. Administration via the i.v. route was used to insure the results do not reflect the ability of compound 9 to cross the gut barrier but rather describe the effective partitioning of the drug once it is in the bloodstream. As shown in (Fig. 7A), compound 9 displayed a dose-response in the writhing assay with a markedly potentEC50 of 0.032 mg/kg. In contrast, no activity could be detected in the hotplate assay with the highest dose of compound 9 tested (30 mg/kg; Fig. 7B). For comparison, morphine at 5 mg/kg will elicit a maximum response after 15 min (shown on the graph), which wanes during the course to the experiment. Using 30 mg/kg as a minimum EC50in CNS-mediated pain, we can calculate a peripheral selectivity for compound 9 of >900-fold.
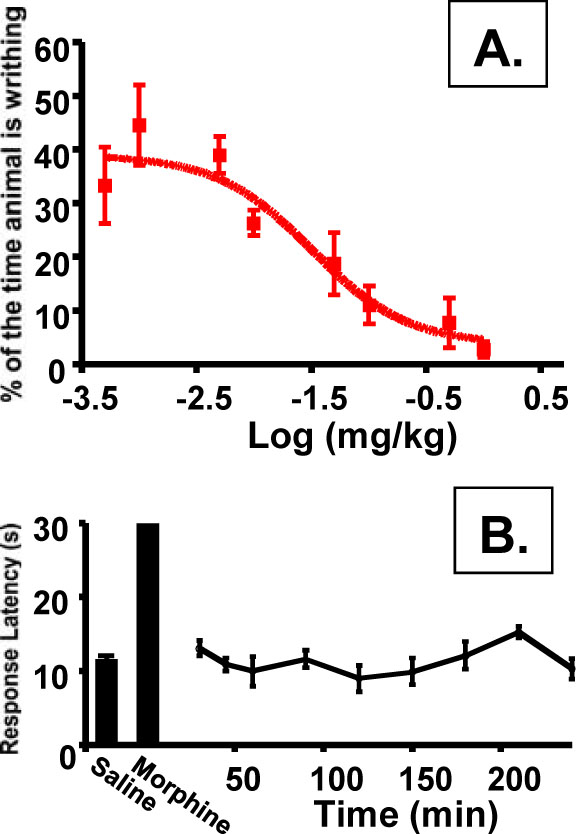
Peripheral selectivity of compound 9. A). i.v dose-response in writhing assay (EC50 = 0.032; 95% confidence level of 0.0095 to 0.1069 mg/kg; Individual points are the mean ± SEM, n = 3 for all data points). B). hotplate assay of compound 9 at 30 mg/kg. Individual points are the mean ± SEM, n= 3 for all data points. No change from saline control was detected.
DISCUSSION
Conventional opioids are the most commonly used treatment for severe chronic peripheral pain, but are less than ideal drugs due to off-target toxicity effects and the potential for development of addiction and/or tolerance. The toxicities result from either lack of receptor subtype selectivity or, more importantly, the ability of opioids to pass into the CNS to target central opioid receptors. Of the three major opioid receptors, recent evidence suggests that kappa-selective agonists have the greatest potential for treating chronic peripheral pain [13,18,20,22]. Non-peptidic kappa selective compounds developed thus far still exhibit undesired effects as the compounds are not peripherally restricted. In contrast, novel peptidic-based kappa selective compounds such as CR665 are peripherally restricted but show little or no activity administered orally [6-8]. This is likely due to a lack of gut barrier permeability and/or extensive degradation by gut peptidases. Oral administration of any analgesic would appear to be a requirement for clinical and out-patient use for the treatment of chronic peripheral pain. In previous work, a compound that is sufficiently active and peripherally restricted when administered orally has not been identified.
A peptide modification platform, in which proprietary non-natural analogs of the cationic amino acids Arg and Lys are synthesized, has been developed and implemented in our laboratory. These modifications are designed to change the PK/PD parameters of the peptides in which they are substituted, often to dramatic effects. Notably, a wide number of bioactive peptide derivatives of the fully active neurotensin(NT) fragment hexapaptide NT(8-13) have been engineered that are able to cross the blood brain barrier (and to some extent, the gut barrier) to promote centrally mediated effects; at this time, compounds are in preclinical development as novel antipsychotics, analgesics and for the treatment of stroke patients [14-16]. For these compounds, analgesic activity is mediated through the agonism of brain NT receptors; the compounds do not bind to activate opioid receptors [unpublished] and peripheral NT receptors are not involved in pain attenuation. For treating peripheral pain, kappa-selective opioid agonists that are orally bioavailable, but centrally restricted, are what is required for a potential pharmaceutical.
The CR665 derivative, compound 9, appears to fulfill these requirements. It binds potently to activate the kappa receptor with an EC50 of 29.9 nM. The agonist selectivity for kappa over mu or delta was minimally >33,400 and likely much greater than that. It has a peripheral versus central selectivity of at least 900-fold, and exhibits oral activity at a reasonably potent EC50 of 4.7 mg/kg. At this point CR665 is not considered a candidate for an oral drug [13] although Cara Therapeutics reports a CR665 derivative, CR845, has approximately 15% oral bioavailability in a Phase 1 clinical trial [13] when included in a formulation to enhance peptide oral activity. It is anticipated that compound 9, or a second-generation compound, will improve upon this without the need for specific formulation. One difficulty in the design of future compounds is that a perceptible structure-activity (SAR) relationship is not apparent for the set of compounds evaluated. This could be due to simply a limited structural data set, or differential accessibility and/or binding of the kappa versus the other opioid receptors (in the latter case, binding clearly must be non-receptor activating).
Of note is that, although 9 is only a first generation compound, the EC50 approaches a druggable level for an oral analgesic. Second-generation compounds are under evaluation to maximize the oral activity, ease of synthesis, and other parameters. While evaluations of potential addiction and tolerance were not performed in this study, the literature suggests the general lack of each for all peripheral kappa receptor agonists [5-7]. In conclusion, it is believed that compound 9, or a subsequent second-generation derivative, is a candidate for development as an analgesic for chronic peripheral pain.
CONFLICT OF INTEREST
The author(s) confirm that this article content has no conflicts of interest.
ACKNOWLEDGEMENT
This work was supported by NIH grants MH-65099 to TAD and NS-073378 to TAD and S.P. Yu, (Emory University), co-PI.
REFERENCES
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]

