All published articles of this journal are available on ScienceDirect.

Roles of PI3K/AKT/PTEN Pathway as a Target for Pharmaceutical Therapy
Abstract
Multiple enzymes participate in the phosphorylation of a group of phosphoinositide lipids. Because of their important role in signal transduction, the dysregulated metabolism of phosphoinositides represents a key step in many disease settings. Loss of their function has been demonstrated to occur as an early event a wide variety of carcinogenesis and has therefore been suggested as a biomarker for the premalignant disease. In addition, genetic alterations at multiple nodes in the pathway have been implicated in several other diseases. Accordingly, given this pervasive involvement in many diseases, the development of molecules that modulates this pathway has been initiated in studies. They have been the focus of extensive research and drug discovery activities. A better understanding of the molecular connections could uncover new targets for drug development.
1. INTRODUCTION
Many pharmacologic inhibitors directed against components of intracellular signaling pathways have been developed to improve therapeutic performance. The phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) / phosphatase and tensin homologue deleted on chromosome ten (PTEN) signaling axis plays a central role in cell proliferation, growth, and survival under physiological conditions [1]. It has now become clear that the PI3K/AKT/mTOR/PTEN signal transduction pathway plays an important role in both normal and malignant cells and thus is an attractive target for targeted molecular therapy. In particular, effective targeting of this PI3K/AKT/mTOR/PTEN signaling network with small pharmacological inhibitors could result in less toxic and more efficacious treatment of cancer patients [2, 3]. In addition, PI3K has been demonstrated to induce expression of the multidrug resistance-associated protein, suggesting that high PI3K activity induces drug resistance [4, 5]. Aberrant regulation of this signaling module has been observed not only in a large group of cancers but also in several other diseases. Because activation of these molecules in the pathway is thought to correlate with poor prognosis and drug resistance, it is considered to be a promising target for therapy [1, 4, 5]. Actually, a number of pharmacologic inhibitors directed against both individual and multiple components of this pathway have already been developed to improve therapy [1-5]. They will be of primary importance in understanding the clinical relevance of PI3K/AKT/mTOR/PTEN modulation in different diseases indications. This review highlights how the PI3K/AKT/mTOR signaling affects survival, proliferation, and drug-resistance of cells including cancer cells. The safety and efficacy of PI3K/AKT/mTOR signaling inhibitors will be also discussed.
2. ROLES OF PI3K/AKT/ PTEN
The PI3K/AKT/mTOR/PTEN signaling pathway is well-known as regulating metabolism, cell growth, and cell survival (Fig. 1). Active form of PI3K is carcinogenic, and mutations of the gene are found in a variety of human cancers [6]. These PI3Ks are heterodimers consisting of a regulatory subunit such as p85 and a catalytic subunit such as p110. Although mutations in PI3K, AKT, or the upstream regulators have been detected in cells from patients with cancers, these mutations appear to be rare [1, 6]. The PI3K forms a family that can be divided into 3 classes based on the structure, distribution, and mechanism of activation [7]. Class I PI3Ks are further divided into class IA and class IB based on their different associated adaptors. Class IA PI3Ks and class IB PI3Ks are activated by receptor tyrosine kinases (RTKs) and by the G-protein-coupled receptors, respectively. The important substrate for these class I PI3Ks is phosphatidylinositol 4,5 bisphosphate [PI(4,5)P2], resulting in the formation of phosphatidylinositol 3,4,5 trisphosphate [PI(3,4,5)P3]. AKT is a downstream target of those PI3Ks. Human AKT has three isoforms: AKT1, AKT2, and AKT3 [8]. The PIP3 leads to the membrane recruitment of the AKT, and binds to phosphoinositide-dependent kinase 1 (PDK1) via their plekstrin homology (PH) domains, then PDK1 phosphorylates AKT in the kinase domain (Thr 308 in AKT1). For the full activation of AKT, the additional phosphorylation (Ser 473 in AKT1) by PDK2 is required [9] (Fig. 2). AKT then moves to the cytoplasm and nucleus, where it phosphorylates several downstream targets to regulate various cellular functions. AKT inhibits the GTPase-activating protein (GAP) activity by phosphorylating tuberous sclerosis complex 2 (TSC2), which leads to activation of mTOR complex [10]. The mTOR mediates the phosphorylation of the ribosomal protein S6 kinases leading to the release of the translation initiation factor eIF4E [11]. The GSK3 is a downstream target of AKT and is also a serine/threonine kinase. GSK3 was originally identified as playing roles in the regulation of glycogen synthesis in response to insulin receptor stimulation [12], which has been shown to be involved in cellular proliferation, apoptosis, and circadian entrainment, in addition to the regulation of glycogenesis [13]. Neuroprotective mechanisms in response to estrogen have been shown to transmit via the GSK3 signaling [14].
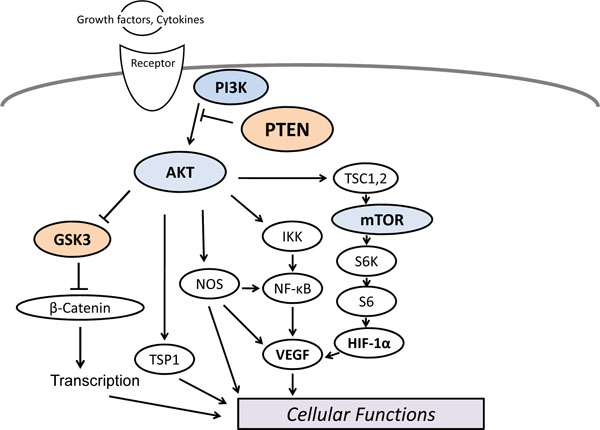
Schematic representation and overview of PI3K/AKT/mTOR/PTEN signaling. Note that some critical pathways have been omitted for clarity.
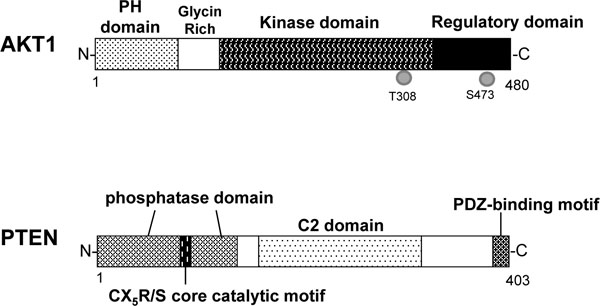
Schematic protein structures of human AKT1 and PTEN. Note that the sizes of protein are modified for clarity. PH domain= pleckstrin homology domain; C2 domain= a protein structural domain involved in targeting proteins to cell membranes; PDZ= a common structural domain in signaling proteins (PSD95, Dlg, ZO-1, etc).
The activity of PI3Ks can be inhibited by PTEN. Schematic structure of the predicted PTEN protein is shown in (Fig. 2). The PTEN has protein phosphatase activity and lipid phosphatase activity [15]. PTEN negatively regulates the PI3Ks and hence the AKT signaling [16]. The structure endows PTEN with its preference for acidic phospholipid substrates such as PIP3. PTEN protein consists of N-terminal phosphatase, and C-terminal C2, and PDZ (PSD-95, DLG1, and ZO-1) binding domains. The PTEN CX5R(S/T) motif resides within an active site that surrounds the catalytic mark with three basic residues, which are critical for PTEN lipid phosphatase activity. The C-terminus of PTEN contains two PEST (proline, glutamic acid, serine and threonine) sequences involved in protein degradation [17]. PTEN can be regulated by post-translational phosphorylation, acetylation, oxidation, and so forth [18]. Tissue-specific deletion of PTEN can result in autoimmunity, glucose dysregulation or neurological deficits, in addition to the carcinogenesis. Further Furthermore, PTEN may be involved in a disease state such as Parkinson’s disease (PD) [19]. Several evidences imply that genes associated with familial PD regulate cell death and/or the cell cycle related to AKT/PTEN pathway [20, 21]. In a mouse model, PTEN deletion in granulosa cells reduces apoptosis and increased cellular proliferation, suggesting that PTEN inhibitor can activate the primordial follicles. Clinical application can be expected the activation of follicles by PTEN inhibitor in ovarian cortical tissues for fertility preservation.
3. PI3K/AKT/MTOR INHIBITORS IN CANCER THERAPY
The PI3K/AKT pathway is frequently activated in human cancers. A variety of signals including growth factors and nutrients leads to the pathway activation. LY294002 and wortmannin are the best characterized PI3Ks inhibitors which prevent ATP to bind to and activate PI3Ks [22]. Both LY294002 and wortmannin induce apoptosis in cancer cells and rescue drug sensitivity. Both inhibitors are low molecular weight compounds and are also cell-permeable. Wortmannin is a natural metabolite and inhibits all class PI3Ks members with a 50% inhibitory concentration (IC50) of 2-5 nM. LY294002 is a flavonoid-based synthetic compound and inhibits PI3Ks with an IC50 of 1-20 μM [23]. LY294002 blocks not only PI3Ks activity but also mTOR to the same extent as PI3Ks. Both wortmannin and LY294002 bind to the p110 catalytic subunit of PI3Ks, leading to the blockade of ATP bound to the active portion. PI3K inhibition with LY294002 is reversible, while wortmannin irreversibly inhibits PI3Ks [22, 24]. Several other PI3K inhibitors have been discovered to affect proliferation and survival of cancer cells [25]. Perifosine is a water soluble synthetic alkylphosphocholine with oral bioavailability, which inhibits AKT phosphorylation through interaction with the PH domain of AKT, resulting in disruption of its PH domain-dependent localization to the cell membrane [26] (Fig. 3). Perifosine reduces cell proliferation and induces apoptosis accompanied by AKT dephosphorylation in a wide variety of cancers [27]. Perifosine also targets the MER/ERK 1/2 pro-survival pathway and activated pro-apoptotic JNK. PI-103 is a synthesized PI3K inhibitor of pyridofuropyrimidine, which synergistically sensitizes leukemia stem cells to daunorubicin-induced cytotoxicity [28]. Furthermore, PI-103 enhances arsenic trioxide cytotoxicity in a heat shock factor 1-dependent manner [29]. KP372-1, an AKT inhibitor, can induce apoptosis in primary leukemic cells and cell lines without affecting the survival of normal hematopoietic progenitors [30]. KP372-1 directly inhibited the kinase activity of AKT and PDK1 in a concentration-dependent manner. Furthermore, KP372-1 decreased the phosphorylation of the S6 ribosomal (Ser240/244) protein [31]. The mTOR inhibitors are the most developed class of compounds which include rapamycin and its derivatives, which bind to FK506 binding protein 12 (FKBP12) [32]. The rapamycin/FKBP12 complex then binds mTORC1 and inhibits downstream signaling [33]. Rapamycin cytotoxicity could be intensely increased by co-treatment with etoposide [34]. Treatment of HL60 cells with phosphatidylinositol ether lipid analogs, a PKB inhibitor, also results in inhibition of proliferation and sensitization to chemotherapeutic agents [35]. ATP-competitive mTOR inhibitors have been generated that inhibit the activity of both mTORC1 and mTORC2 [36]. Compared with rapamycin, the mTORC 1/2 inhibitor PP242 more efficiently reduces the development of leukemia in mice [37]. Combining the PI3K/PDK1 inhibitor BAG956 with RAD001 also results in a synergistic reduction in tumor growth [38]. PKI-587, a class I PI3Ks inhibitor suppresses phosphorylation of PI3K/AKT/mTOR effectors and induces apoptosis in cancer cells [39]. NVP-BEZ235, an orally bioavailable imidazoquinoline derivative that inhibits the activity of both PI3K and mTOR by binding to their ATP-binding pocket, reduces proliferation and survival in leukemic cell lines without affecting normal hematopoietic progenitors [40] (Fig. 4). However, the administration of PI3K/AKT/mTOR inhibitors can give rise to a potentially life-threatening adverse effect such as pneumonitis and so on [41].
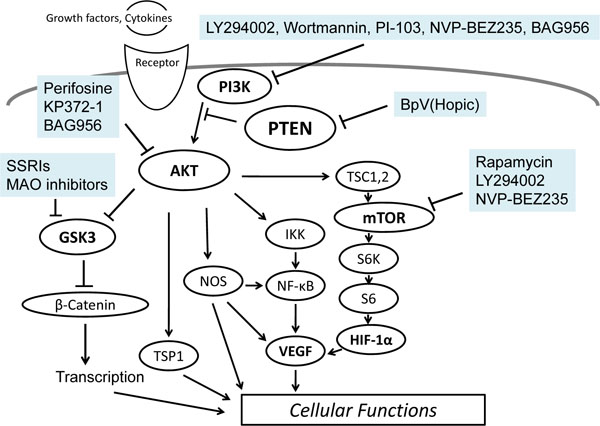
Several inhibitors linked to the PI3K/AKT/mTOR/PTEN signaling pathway are shown. Arrowhead means stimulation whereas hammerhead represents inhibition, suggesting implication of PI3K/AKT/mTOR/PTEN modulators for pharmaceutical therapy of various diseases.
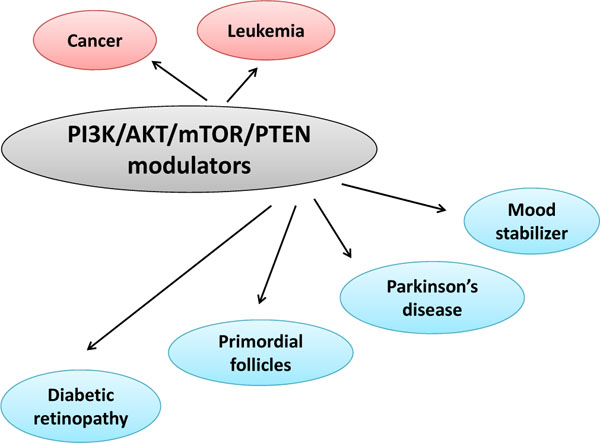
4. PI3K/AKT/PTEN INHIBITORS IN THERAPY AGAINST OTHER DISEASES
Some kind of psychoactive drugs have been shown to modulate the activity of the AKT/GSK3 signaling (Fig. 3). It has been reported that antidepressants acting on serotonin neurotransmission activate AKT and inhibit GSK3 [42, 43]. AKT has a lot of substrates including the GABA receptor [44]. Inhibition of AKT in neurons may increase excitability through reductions in GABA neurotransmission [45]. Interestingly, drugs like SSRIs and MAO inhibitors that elevate serotonin synaptic transmission have been shown to inhibit the GSK3 [46]. In contrast, drugs that raise dopamine neurotransmission decrease the inhibitory phosphorylation of the GSK3 [47]. Typical antipsychotics can prevent the inhibition of AKT by activation of the GSK3 through blocking dopamine D2 receptors [48]. Atypical antipsychotics may also affect the regulation of the GSK3 [49]. Actually, such regulation between AKT and GSK3 has been reported after treatment with haloperidol [50]. Remarkably, lithium activates PI3K itself, which in turn results in activation of the AKT, then phosphorylation and inactivation of the GSK3 [51]. Caspase-3 activation in apoptosis pathways and glutamate-induced reduction of AKT activity in addition to the associated neuronal toxicity are prevented by the lithium treatment [52]. On the contrary, PTEN inhibition reduces neurological damage after ischemic brain injury. The pharmacological inhibition of PTEN protects against brain injury in a dose-dependent manner and the protective effect might be induced through upregulation of the PI3K/AKT pro-survival pathway, suggesting a new therapeutic strategy for neuro-protection. Valproate, a mood stabilizer, has been reported to inhibit the GSK3 [53]. Furthermore, direct inhibition of GSK3 isoforms has similar effects to some of those of antidepressants [54]. Guanosine increases AKT and GSK3 phosphorylation, suggesting that this pathway plays an important role in the neuro-protective effect [55]. The neuro-protective effect of guanosine is abolished by blocking the AKT pathway with LY294002 [56]. Accumulative evidences pointing to PI3K/AKT/GSK3 modification in depression provide a novel implication of the antidepressant mechanisms.
The PI3K/AKT/GSK3 activation is maintained by extracellular signals, and it maintains the survival of cells. For example, the survival of primordial follicles is maintained through the PI3K pathway [57]. Absence of PTEN also increases PIP3 that leads to active AKT signaling, resulting in the maintenance of the primordial follicles. From the studies of PTEN-/- mice, PTEN is crucial for the maintenance of inactivity of primordial follicles [58]. The mTOR deletion is expected to accelerate the loss of primordial follicles. So, PTEN inhibitor, such as BpV(Hopic), has been shown to effectively activate primordial follicles in ovarian cortical tissues. Short-term treatment of mouse ovaries with PTEN inhibitor increases the activation of primordial follicles and the number of follicles [59]. The follicular activation with PTEN inhibitor could be applied to generate more germ cells. PTEN inhibitors might have clinical potential for generating fertilizable oocytes. The mTOR may regulate protein synthesis and cell growth in the survival of primordial follicles.
Prosurvival PI3K/AKT and p70 S6K signaling is diminished in models of diabetic retinopathy (DR) [60]. However, inhibitors of PI3K/AKT/mTOR pathway may present a unique opportunity for the management of DR. Some mTOR inhibitors seem to be efficacious in managing the DR, which suppress HIF-1α, VEGF, and breakdown of the blood-retinal barrier. Those mTOR inhibitors initiate an inhibitory effect on inflammation influencing the progression of the DR. Some of growth factors are known to play major roles in the induction of angiogenesis depend on PI3K/AKT/ mTOR for prolonging the cell survival signals that are operant in pathological angiogenesis. DR might stem from changes in the PI3K/AKT pathway. Accordingly, the inhibition of PI3K/AKT/ mTOR pathway could have beneficial therapeutic effects for the management of proliferative DR [61].
5. PERSPECTIVE
An important mediator implicated in regulation of several diseases is the PI3K/AKT/ PTEN signaling segment. In particular, because activation of this signaling pathway has been demonstrated to induce malignancies and is thought to associate with poor prognosis and enhanced drug resistance, it is considered to be a promising target for therapy. In addition, the signaling pathway appears to be highly deregulated in a large number of patients with above mentioned diseases. Efforts to exploit pharmacological inhibitors of the cascade which show efficacy and safety in the clinical setting are underway. It is unlikely that inhibition of a single signaling pathway will achieve cure in each diseases. However, combining inhibitors with conventional chemotherapy drugs could be an effective therapeutic option for the patients. Some of human diseases including cancer are known to evolve through a multistage process which can extend over a period of several years. Therefore, they progressively accumulate mutations and epigenetic anomalies in expression of multiple genes. As a consequence, disorders are characterized by multiple signaling abnormalities and the deregulated pathways may be redundant. It could be difficult to find the right combinations of targets. Possible precise involvement of the PI3K/AKT /GSK3/mTOR/PTEN in signaling induced by several diseases has remained unexplored. Although further research is required to examine the safety and efficacy of the inhibitors, these compounds appear to possess promising therapeutic activities. It is probable that inhibition of the signaling in definite disease populations could be associated with good outcomes. In this review, the efficacy of the signaling inhibitors has been discussed as well as the potential of combination therapy with inhibitors focused against related signal transduction molecules. Understanding of the intracellular mechanisms could provide innovative insights into the development of new therapeutic approaches. To further optimize therapeutic regimens, research should also be focused on combination of inhibitors of the PI3K signaling with inhibitors directed against other signal transduction molecules. Further research is warranted to examine the safety and efficacy of these regimens.
CONFLICT OF INTEREST
The author(s) confirm that this article content has no conflicts of interest.
ACKNOWLEDGEMENT
This work was supported by grants-in-aid from the Ministry of Education, Culture, Sports, Science and Technology in Japan. In addition, this work was supported in part by the grant from Nakagawa Masashichi Shoten Co., Ltd.
ABBREVIATIONS
REFERENCES
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link] [PMC Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link] [PMC Link]
[PubMed Link]

