All published articles of this journal are available on ScienceDirect.

Docking Studies of Methylthiomorpholin Phenols (LQM300 Series) with Angiotensin-Converting Enzyme (ACE)
Authors Info & Affiliations
Abstract
A main target in the treatment of hypertension is the angiotensin-converting enzyme (ACE). This enzyme is responsible for producing angiotensin II, a potent vasoconstrictor. Therefore, one of the targets in the treatment of hypertension is to inhibit ACE activity. Hence, this study’s aim is to use computational studies to demonstrate that the proposed heterocyclic compounds have a molecular affinity for ACE and that, furthermore, these heterocyclic compounds are capable of inhibiting ACE activity, thus avoiding the production of the vasopressor Angiotensin II. All this using computer-aided drug design, and studying the systems, with the proposed compounds, through molecular recognition process and compared with the compounds already on the market for hypertension.
INTRODUCTION
In recent years, cardiovascular diseases have become a serious problem worldwide. The World Health Organization has reported an increase in the number of patients suffering from this disease. Currently, existing treatments for high blood pressure are not very effective and are generally uncomfortable for patients. This relies in that the patient need to have a very strict control in the dosage and in the moment of the administration of the drug. And also some patients have an unfavorable response after the administration, leading them to a fast blood pressure reduce. One of the most widely used compounds for the treatment of hypertension is captopril. Like many others on the market, this drug was designed with computational tools. Over the last few decades, computational studies, together with rational drug design, have become a critical part in the development of new drugs. Currently, cardiovascular diseases are a serious health problem worldwide. One example of cardiovascular disease is arterial hypertension, which is defined as increased systolic pressure, diastolic pressure, or both. Hypertension is a silent, asymptomatic disease, and as a result, not many people know that they suffer from it. Hypertension is an important risk factor, contributing to other cardiovascular diseases such as blood vessel disorders, coronary heart disease, aortic aneurysm, stroke, etc.
Cardiac arrhythmia is another cardiovascular problem. An arrhythmia is any disorder of the heart rate that may cause stroke. Heart rhythm disorders may be caused by genetic factors or occur when the heart muscle (myocardium) is damaged, sometimes by hypertension.
In 1985, Stout and colleagues conducted studies on the compound changrolin [1], which has antiarrhythmic and hypotensive effects, in order to identify which region of the changrolin molecule was responsible for biological activity. Their studies indicated that the biological activity of changrolin relies on region II in this molecule (Fig. 1) [2, 3].
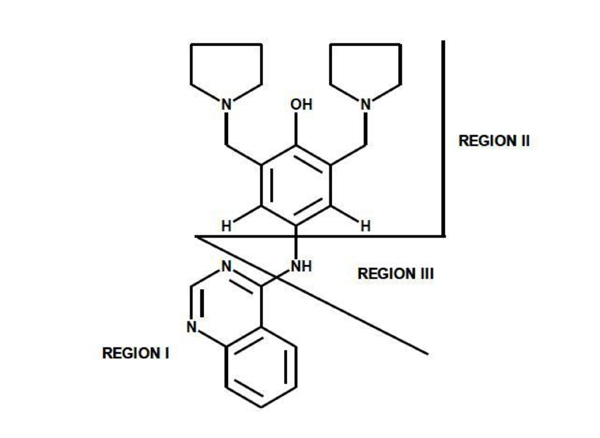
Changrolin regions studied by Stout et al.
Our group of Medicinal Chemistry at FESC-UNAM has reexamined Stout’s investigations by studying and evaluating changrolin analogues in which the pyrrolidinyl groups are replaced with thiomorpholine. In 2007, Velázquez Sánchez et al. synthesized and characterized a series of compounds derived from phenol, called LQM300 [4], with possible antihypertensive activity (Fig. 2). Aguilar Martínez and her research group studied these compounds in vivo and found that these phenol derivatives had an antihypertensive effect [5]. Moreover, they suggested the inhibition of the angiotensin-converting enzyme as a possible mechanism of action.
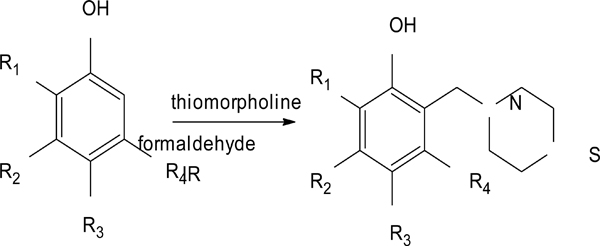
Synthesis pathway of the LQM300 series.
According to preliminary results on the antihypertensive effect of the LQM300 series shown in (Table 1), the compounds showed higher activity than captopril and enalapril in vivo (unpublished results). Therefore, the objective of this study is to determine the possible interaction of the LQM300 compounds with the human angiotensin-converting enzyme. Furthermore, using molecular recognition studies, to identify the best candidates for this interaction.
LQM Compounds with Structures and Melting Points
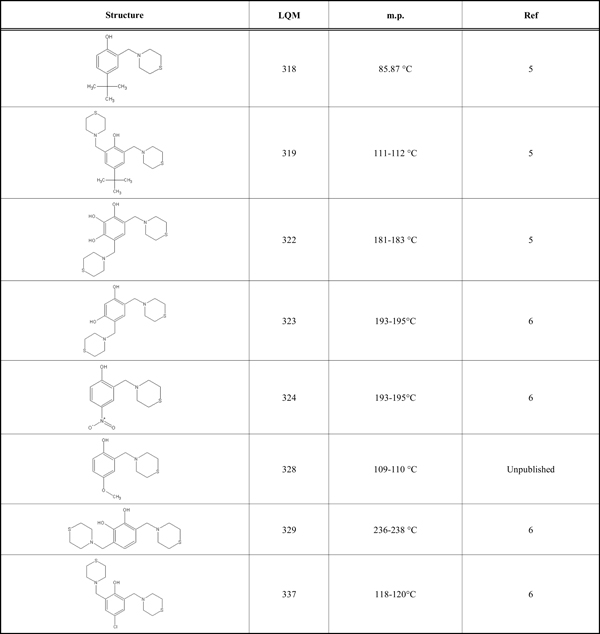 |
MATERIAL
This study used the Molecular Operating Environment (MOE) software, version 2012.10, installed on a Mac OS X Quad-Core Intel Xeon with 32 gigabytes of RAM and two 2.8 GHz processors. The structure of the human angiotensin-converting enzyme was downloaded from the Protein Data Bank [15] with PDB ID 1UZE. This structure is an ACE-enalaprilat complex with a resolution of 1.82 Å. The LQM300 compounds are phenol derivatives; their structures are shown in (Table 1) [6]. All compounds have been reported previously, including their corresponding structures, with the exception of LQM328. These compounds were modeled and optimized with the MOE, Gaussian 09 and Gaussview programs, with the protocols for each described in the methods section. The Gaussian 09 and Gaussview [7] software are installed on a Linux computer (CentOS) with 24 Gigabytes of RAM and 24 processors.
METHODS
The angiotensin-converting enzyme was downloaded from the Protein Data Bank (http://www.pdb.org/pdb/ home/home.do) with PDB ID 1UZE. The protein was analyzed using the MOE correction command “Structure Preparation” in order to perform a 3D protonation. This command helped to produce the right geometric orientation of the hydrogen atoms present in the enzyme and in the environment. This calculation was performed using the MMFF94x force field, which is parameterized for this kind of system with large proteins and small organic molecules.
Once obtained the proper orientation of the hydrogen atoms in the enzyme and the environment, the protein was saved under the PDB extension to maintain proper connectivity and guidance after the 3D protonation calculation. After the protonation, the possible ACE interaction sites were studied using the "Site Finder" command, which computes the possible recognition sites from the 3D atomic coordinates of the receptor. The Site Finder command is considered a geometric method, as no energy models are used. Instead, the relative positions and accessibility of the receptor atoms are considered along with a rough classification of chemical types. Once these regions were calculated, dummy atoms were assigned to these sites and later used to make the molecular docking calculation for specified sites.
The LQM300 molecules were modeled using the "Builder" command on the MOE interface. The molecular modeling was done while trying to maintain the correct geometric shape and stereochemistry in order to avoid convergence errors when performing the geometric optimization. The geometry optimizations of the LQM300 compounds were performed using stochastic optimization. The parameters of the conformational analysis protocol were set as follows: rejection limit of 100 molecules, iteration limit of 100,000 and default parameters for the rest. Once obtained the geometric optimizations, these structures were validated with Gaussian 09 software. Two kinds of calculations were made to perform the validation step: one was an energy calculation without geometry optimization (single point calculation), and the other was an energy calculation with full geometry optimization. These calculations were done to compare the structure resulting from a molecular mechanical optimization against the final optimized structure from a quantum mechanical calculation. For the quantum mechanical calculation, the hybrid B3LYP from the Density Functional Theory (DFT) was used with a 6-31+g(d,p) level of theory and a PCM model with water as the solvent. For all the quantum mechanical calculations, molecular frequencies were also computed.
Prior to the molecular recognition between the LQM300 compounds and ACE, the MOE docking protocol was validated for this enzymatic system. To achieve this, enalaprilat was extracted from the original PDB file and then reinserted using the rigid-flexible docking protocol. For this protocol used the following parameters were used: "triangle matcher" for placement, and the "London dG" function for rescoring, with a final retention of 30 complexes. Finally, for the flexible-flexible docking protocol, the parameters mentioned above were used, with the addition of a refinement of the resulting complexes with the force field (MMFF94x) and London dG for a second rescoring.
Once the MOE docking protocol was validated, molecular recognition calculations near where the enalaprilat was originally interacting were performed, as Martínez A. [6] found that the action mechanism of the LQM300 compounds in vivo is very similar to that of ACE inhibitors such as enalaprilat. Finally, the protein interactions were computed with all the ligands using Protein-Ligand Interaction Fingerprints (PLIF) to determine which amino acids and water molecules were important during the molecular recognition process. Also all these interactions were compared of the LQM300 compounds to those reported in the PDB for ACE interactions with the 1UZE complex and with the structure with captopril as a ligand (PDB ID 1UZF).
RESULTS AND DISCUSSION
Chemistry of compound LQM328. The methodology of synthesis has been described by Velázquez et al. [5,6].
4-methoxy-2-(thiomorpholin-4-ylmethyl)phenol, m.p.109-110 °C. Yield 30%. Reaction time: 16 minutes.
IR (cm-1; CHCl3 pellet) 3298, 3030, 2910, 1493. 1H-NMR (300 MHz; CDCl3; Me4Si, δH): 10.085 (1H, s,OH), 6.75 (2H, m), 6.55 (1H, m), 3.74 (3H,s), 3.67 (2H), 2.83 (4H,m), 2.72 (4H,m). 13C-NMR (δC):152.57, 152.31, 121.4, 116.54, 114.65, 113.68, 62.33, 55.74, 54.43, 27.92. FAB-MS (M+1) 240 (8%), 224 (21%), 137 (100%). Calculated for C12H17NO2S. C 60.20%; H 7.19%; N 5.90%; O 13.32%; S 13.36%.
The resulting 3D molecular structures of the LQM300 compounds following the stochastic conformational analysis calculation are shown in (Fig. 3).
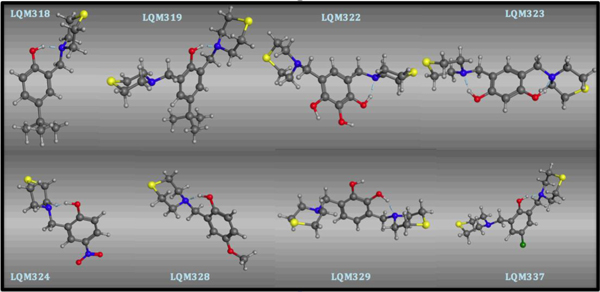
Structural conformation of the LQM300 compounds at the end of the stochastic minimization.
A quantitative comparison between the molecular mechanical and quantum mechanical methods of optimization is presented in (Table 2). There are two values in this comparison: the MM box, which in all cases is zero because the molecular mechanics structures with themselves were compared just to confirm that the comparison protocol was correct, and the values in the QM box, which indicate the difference between the final molecular mechanics optimization and final quantum mechanical optimization. In all cases, the difference was greater than 2 Å, indicating that the structures are geometrically different. However, the function of the geometry optimization in Gaussian 09 converged much faster when we used molecular mechanics first than when we used only Gaussview and Gaussian 09. Therefore, the time needed for the calculation was reduced when optimizing the geometry first, followed by the quantum level optimization. This helps us have a lower energy structure with a high level of theory while maintaining a relatively low computational cost. Thus, the structures used for molecular recognition were those that had first been optimized by molecular mechanics, followed by quantum mechanics.
Comparison Between the Final Optimization of the Molecular Mechanical Structure (MM) and the Quantum Mechanical Structures (QM Opt), Using the RMSD (Root Mean Square Deviation)
| LQM318 | RMSD | LQM324 | RMSD |
| MM | 0.0000 | MM | 0.0000 |
| QM Opt | 2.6508 | QM Opt | 2.7895 |
| LQM319 | RMSD | LQM328 | RMSD |
| MM | 0.0000 | MM | 0.0000 |
| QM Opt | 4.5736 | QM Opt | 3.9047 |
| LQM322 | RMSD | LQM329 | RMSD |
| MM | 0.0000 | MM | 0.0000 |
| QM Opt | 3.7984 | QM Opt | 4.7169 |
| LQM323 | RMSD | LQM337 | RMSD |
| MM | 0.0000 | MM | 0.0000 |
| QM Opt | 5.1089 | QM Opt | 3.7127 |
The validation process consisted of extracting the protein ligand and determining whether the program was able to reinsert the ligand in the same place. The resulting validated structure was compared with the experimental structure reported in the PDB, and finally, the RMSD was calculated to determine if the program can do an adequate reinsertion of the ligand into the protein. The structure resulting from the validation process is shown in (Fig. 4). In this figure, the ligand in green (experimental structure) is inside the ACE pocket, and the blue structure (calculated theoretically) has a very similar arrangement in the same place. Fig. (5) also shows both ligands, but in this case the protein is represented as a tube. We can also observe a small deviation in the protein backbone, but in both figures, the ligand and protein are configured in a very similar manner.
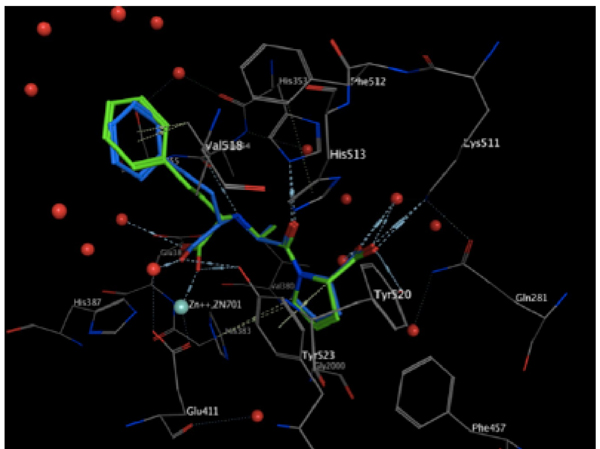
Resulting structure from validation in the pocket. The green ligand is from the experimental structure, and the blue is from the calculated structure.
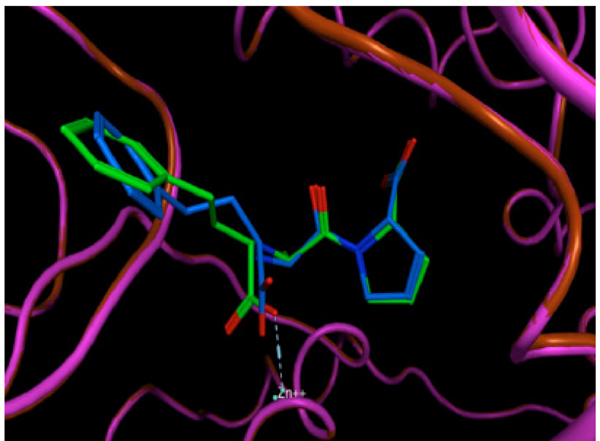
The same resulting structure as in Figure 4, but with the protein represented as a tube.
The RMSD calculation was performed using these structures to determine quantitatively the deviation of the atoms’ location in one structure compared to the other. The comparison is shown in (Fig. 6), represented as a matrix calculation comparing the two structures. The final RMSD value is 0.032 Å. This value indicates that the difference between the two molecular complexes is not significant; therefore, the molecular recognition protocol in the MOE program is reliable for docking studies between ACE and LQM300 compounds.
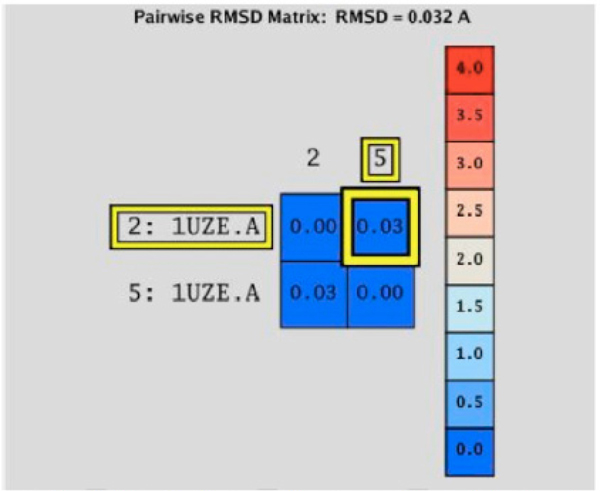
RMSD matrix calculation between the experimental structure (2:1UZE.A) and the calculated structure (5:1UZE.A).
At this point, was concluded that the MOE program was able to reinsert the ligand enalaprilat from the experimental structure. To obtain reliable results, this process must be performed with every system under study, in order to determine the optimal conditions for each and every molecular recognition process [8]. In this case, the MOE program was able to reintegrate the enalaprilat into the ACE with a deviation of 0.032 Angstroms. This deviation suggests that the protocol that was used for the systems is optimal, because it can describe structures favorably.
For molecular recognition between the ACE and the LQM300 compounds, the action was confirmed site by calculating it with the "Site Finder" command in MOE (Fig. 8). This helped us to set an initial position for the LQM300 compounds at the action site with an initial orientation. As shown in (Fig. 7), the enalaprilat (green) is positioned in the pocket, and the spheres indicate hydrophobic centers (red) and hydrophilic centers (white). This calculated recognition site and these centers were used to assign an initial orientation for all the compounds studied.
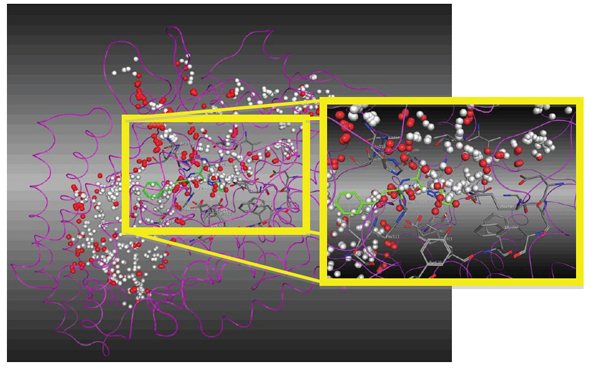
Magnification of the ACE active site with enalaprilat oriented according to the hydrophobic (red) and hydrophilic spheres (white).
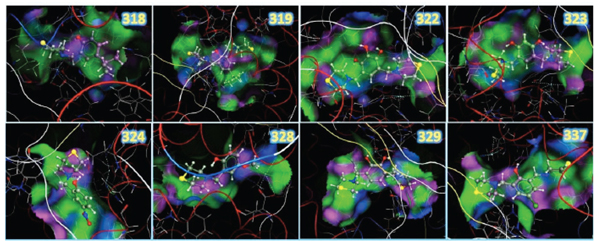
Resulting structures from the docking study between the LQM300 compounds and ACE, showing the zinc, the backbone in tube representation and the pocket surface.
Once the optimal conditions were found, the dummy atoms were calculated to assign an initial position to the LQM300 compounds. The MOE program creates these dummy atoms when hydrophobicity or hydrophilicity is assigned to the molecular space. All these processes depend on the environment inside the pocket and the orientations of the side chain of the amino acids. LQM300 compounds showed favorable orientations within the pocket. During the whole process, any intermolecular collisions were not observed, which would invalidate the molecular recognition process. If these collisions were found in our resulting systems, the results obtained would be less favorable [9].
The molecular recognition between ACE and the LQM300 compounds was performed taking into account the previous validation methodology for this system. In this calculation, the water molecules present in the pocket were took into account, as other studies have established the relevance of these structures for molecular recognition [10]. The final configurations after applying the docking protocol are shown in (Fig. 8). These images show the 3D structures of the LQM300 compounds in the ACE active site, along with the zinc that was visually deployed to corroborate its proximity to the LQM300 compounds.
The resulting structures were analyzed as shown in (Fig. 9). They showed a favorable interaction, as there was neither steric hindrance nor an incorrect geometric arrangement. All LQM300 compounds were coupled within the pocket, and they presented a spatial arrangement that allowed long-distance interactions with ACE.
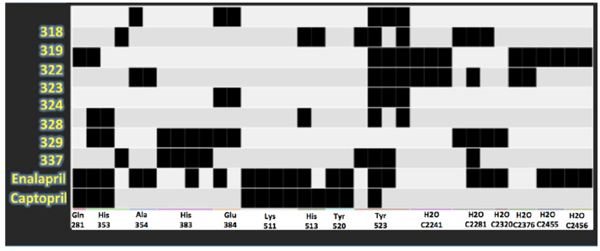
Protein-ligand interaction fingerprints between LQM300, enalaprilat and captopril compounds and ACE. Each dark spot indicates an interaction.
Table 3 shows the calculated energies in kcal/mol for each complex studied with molecular recognition. Fig. (9) shows the main interactions of the LQM300 compounds with ACE using the so-called protein-ligand interaction fingerprints, which helps to observe relevant interactions during molecular recognition between the LQM300 compounds and ACE.
Affinity Energy Computed for the LQM300-ACE Complexes
| Ligand | Affinity energy (kcal/mol) |
|---|---|
| LQM318 | -9.18 |
| LQM319 | -12.85 |
| LQM322 | -9.64 |
| LQM323 | -10.75 |
| LQM324 | -8.27 |
| LQM328 | -7.62 |
| LQM329 | -11.68 |
| LQM337 | -6.94 |
In Fig. (9), the interactions that are present in the experimental data for enalaprilat and captopril were compared. This PLIF table shows that many interactions are also conserved for the LQM300 compounds, which suggests that there are some residues involved during the molecular docking that are critical for the molecular recognition process, such as Tyr 523, Glu 384, His 383, His 353 and Gln 281. In contrast, Lys 511 is only involved in the interactions between ACE and captopril and enalaprilat. The PLIF can also be observed in (Fig. 10), which shows the possible interactions between the ligands and ACE from the perspective of the ligand. As with the PLIF table, these representations show both long-range interactions and direct interactions, according to the relative intermolecular proximity.
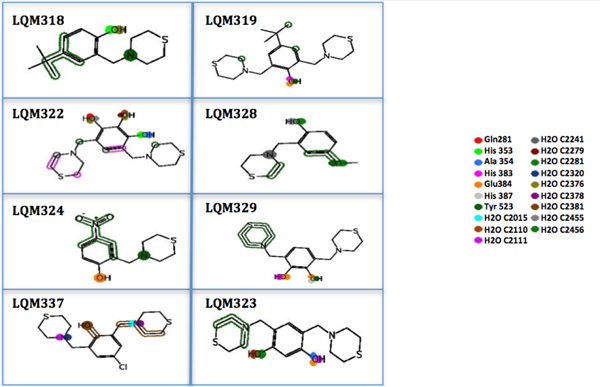
The PLIF from the ligand view shows both long-range interactions and relative LQM300-ACE intermolecular proximity.
The energy results from the experimental data, which were used for comparison in this study, are shown in (Table 4). In this case, the compounds compared were the antihypertensive drugs captopril and enalaprilat. Both molecular complexes were obtained from the Protein Data Bank [8], along with all the structural details on the binding of captopril and enalaprilat to human testicular angiotensin I-converting enzyme [11]. Table 4 shows the energies of the compounds reported experimentally, computed with the MMFF94x force field. These results were calculated using the same protocol, but without any optimization. Fig. (11) shows the PLIF of the experimental ligands in the LQM300 compounds.
Computed Energies for the Experimental Structures without Optimization.
| Ligand | Affinity energy (Kcal/mol) |
|---|---|
| Enalaprilat | -17.34 |
| Captopril | -9.66 |
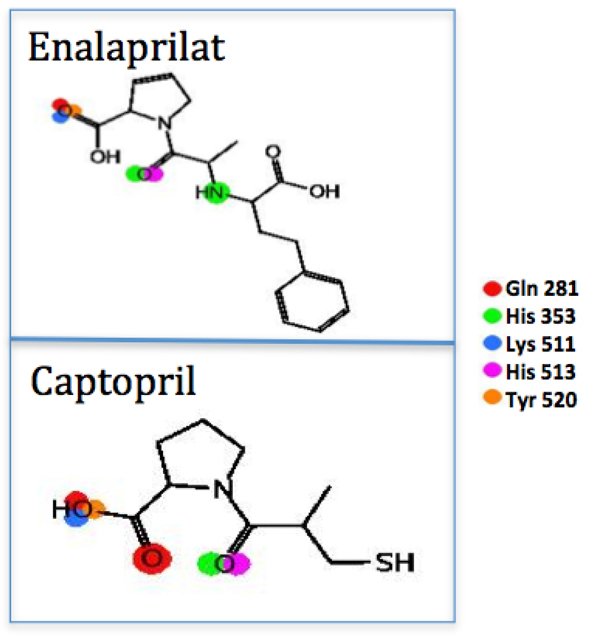
The PLIF from the ligand view for captopril and enalaprilat.
A comparison of the structures of the experimental complexes with LQM300 compounds indicates that the most energetically stable complex is the one with enalaprilat, and this compound has the same interactions with ACE as captopril. However, enalaprilat is the active metabolite of enalapril. It is the first dicarboxylate-containing ACE inhibitor and was developed partly to overcome some of the limitations that captopril presents. The sulfhydryl moiety was replaced by a carboxylate moiety, but additional modifications to its structural design were required in order to achieve a similar potency to captopril.
Enalaprilat, however, has a problem of its own. The structural modifications have been shown to produce unfavorable ionization characteristics that lead to insufficient potency for oral administration (in tablets), making enalaprilat only suitable for intravenous administration. [12, 13]
The other experimental compound that was used for comparison was captopril. This compound demonstrated a theoretically higher energy than both enalaprilat and the compounds LQM319, LQM323 and LQM329. As mentioned above, it was shown that interactions with ACE are the same for captopril and enalaprilat, but molecular recognition showed that there is a difference in the affinity energy. Some of captopril’s disadvantages include the adverse drug reaction (ADR) profile, with cough being the most common ADR. However, captopril is also commonly associated with rash and taste disturbances (metallic taste or loss of taste), which are attributed to the unique sulfhydryl moiety. Captopril also has a relatively poor pharmacokinetic profile, since the short half-life in blood generates the need for two or three doses per day, which may reduce patient compliance [14]. Captopril and enalaprilat compounds showed a direct interaction with or a significant approach to ACE residues Gln 281, His 353, Lys 511, His 523 and Tyr 520. While LQM300 compounds had interactions with Gln 281 and His 353, they also had interactions with Tyr 523, Glu 384 and His 383. The LQM324 compound also presented an interaction mediated by a water bridge; however, this compound was shown to have an activity no more favorable than the experimental compounds.
CONCLUSIONS
The study of molecular recognition between the LQM300 compounds and angiotensin-converting enzyme was performed after the proposed methodology was validated for the system. With this validation was confirmed that the Molecular Operating Environment program is able to reintegrate a ligand extracted from an experimental molecule, with the resulting positions showing a very low level of error in terms of the implicit error of the experimental molecule.
The molecular recognition study with the series of LQM300 compounds showed that the affinity levels were not superior with respect to enalaprilat. In contrast, the LQM319, LQM323 and LQM329 molecules had greater affinity for ACE than captopril, and the LQM300 molecules have highly similar PLIFs with different residues of the protein, while there are slight differences when compared with captopril and enalaprilat. Despite its high affinity for ACE, enalaprilat is a drug that has major drawbacks for administration in hypertensive patients. The methodology proposed here can also be applied to new proposed compounds in the same family, as the validation results described above demonstrate that this methodology is reliable.
Finally, it was determined that the family of LQM300 compounds acts on the angiotensin-converting enzyme as described by Martínez et al., and the final computed energy system suggests that the interaction is very favorable. Therefore, we can offer this group of compounds as candidates to be used as an alternative treatment for hypertension, and, based on their effect on ACE, we can assert that the synthesis, reaction yields and easy purification could give rise to benefits in the treatment of cardiovascular diseases.
This study was supported by PAPIIT/UNAM Projects No. IT223311, IT223011, IG200813 and IT202412. This work was conducted as part of the FESC-UNAM-2012 Project Cátedra: Diseño de Sustancias Bioactivas. E. Angeles would like to thank F. Sotres, M. Hernández, D. Jiménez and R. Valadez for the technical assistance from FESC-UNAM.
CONFLICT OF INTEREST
The author(s) confirm that this article content has no conflicts of interest.
ACKNOWLEDGEMENT
Declared none.
REFERENCES
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]

