All published articles of this journal are available on ScienceDirect.

Design, Synthesis and Biological Evaluation of N4-Sulfonamido-Succinamic, Phthalamic, Acrylic and Benzoyl Acetic Acid Derivatives as Potential DPP IV Inhibitors
Authors Info & Affiliations
Abstract
As incidence rate of type II diabetes mellitus continues to rise, there is a growing need to identify novel therapeutic agents with improved efficacy and reduced side effects. Dipeptidyl peptidase IV (DPP IV) is a multifunctional protein involved in many physiological processes. It deactivates the natural hypoglycemic incretin hormone effect. Inhibition of this enzyme increases endogenous incretin level, incretin activity and should restore glucose homeostasis in type II diabetic patients making it an attractive target for the development of new antidiabetic drugs. One of the interesting reported anti- DPP IV hits is Gemifloxacin which is used as a lead compound for the development of new DPP IV inhibitors. In the current work, design and synthesis of a series of N4-sulfonamido-succinamic, phthalamic, acrylic and benzoyl acetic acid derivatives was carried out. The synthesized compounds were evaluated for their in vitro anti-DPP IV activity. Some of them have shown reasonable bioactivity, where the most active one 17 was found to have an IC50 of 33.5 μM.
INTRODUCTION
Nowadays, diabetes is considered a recognized epidemic that affects large number of people in different societies [1]. The majority have type II diabetes, which is typically associated with being overweight or obese. Type II diabetes is usually treated through lifestyle and oral hypoglycemic agents [2]. The management of diabetes highlights the benefits of controlling blood glucose as near to normal as possible [2,3]. This delays the onset and reduces the severity of microvascular and neuropathic complications [4].
Dipeptidyl peptidase IV (DPP IV) is an aminopeptidase that exists as a free circulating enzyme and bound at the surface of endothelia and other epithelial cells in most tissues, especially in the intestinal mucosa [5]. It cleaves the N-terminal dipeptide from a range of peptides that have either an alanine or a proline residue penultimate to the N-terminus, especially glucagon-like peptide-1 (GLP-1) and gastric inhibitory peptide (GIP). Other potentially affected peptides by DPP IV include gastrin releasing peptide, enkephalins, bradykinin, insulin-like growth factor-1, growth hormone releasing hormone, neuropeptide Y, peptide YY, 1-36 prolactin, substance P and the alpha chains of thyrotropin, luteinizing hormone and chorionic gonadotropin [6].
The antidiabetic effect of DPP IV inhibition was underlined by studies in DPP IV knockout and mutant rodents.
Lack of DPP IV resulted in increased levels of incretin hormone and insulin, with improved glucose tolerance and resistance to diet-induced obesity [7,8]. Preclinical studies with chemical inhibitors of DPP IV demonstrated improved glycemic control in diabetic animal models [9,10]. Several DPP IV inhibitors, ‘gliptins’, have proceeded in clinical development [10,11].
Discovery and optimization of new DPP IV inhibitors as leads for new hypoglycemic agents for treatment of type II diabetes was carried out by several researchers [12,13], resulting in a number of reversible and irreversible DPP IV inhibitors such as Vildagliptin, Sitagliptin, Saxagliptin and Gemifloxacin (Fig. 1) [11,14-18]. DPP IV inhibitors have several advantages over classical antidiabetic agents, such as their compliance for oral administration in a once daily dose [15], causing less risky hypoglycemia, and promoting regeneration and differentiation of β-cells [19,20].
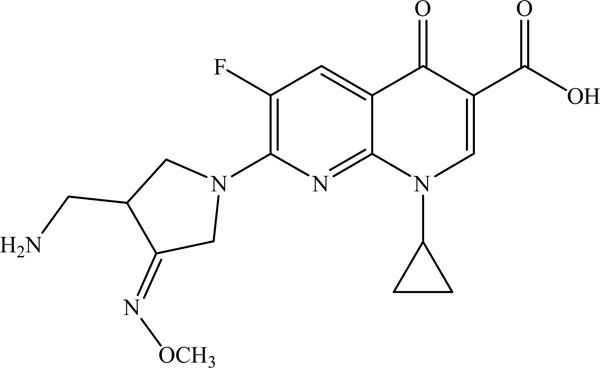
Structure of Gemifloxacin.
As incidence rate of type II diabetes mellitus continues to rise, there is a growing need to identify novel therapeutic agents with improved efficacy and reduced side effects. A great number of research articles and patents have emerged recently dealing with this matter [12,13,21,22].
Earlier, the pharmacophoric space of DPP IV was explored using a set of 358 known inhibitors. A ligand-based three-dimensional (3D) pharmacophores (Hypo4/10, Fig. 2a and Hypo32/8, Fig. 3a) integrated within self-consistent QSAR model for DPP IV inhibitors was developed, suggesting the existence of at least two distinct binding modes accessible to ligands within the DPP IV binding pocket [17, 18]. The pharmacophore models (Hypo4/10 and Hypo32/8) were used as 3D search queries to mine 3D libraries for new DPP IV inhibitors, while the QSAR model predicted their biological activities and therefore prioritize them for in vitro evaluation [17, 18]. The use of this inventive approach was previously reported in the discovery of new inhibitory leads against cholesteryl ester transfer protein [23-25], β-D-glucosidase [26], β-D-galactosidase [27] and N-myristoyl transferase [28].
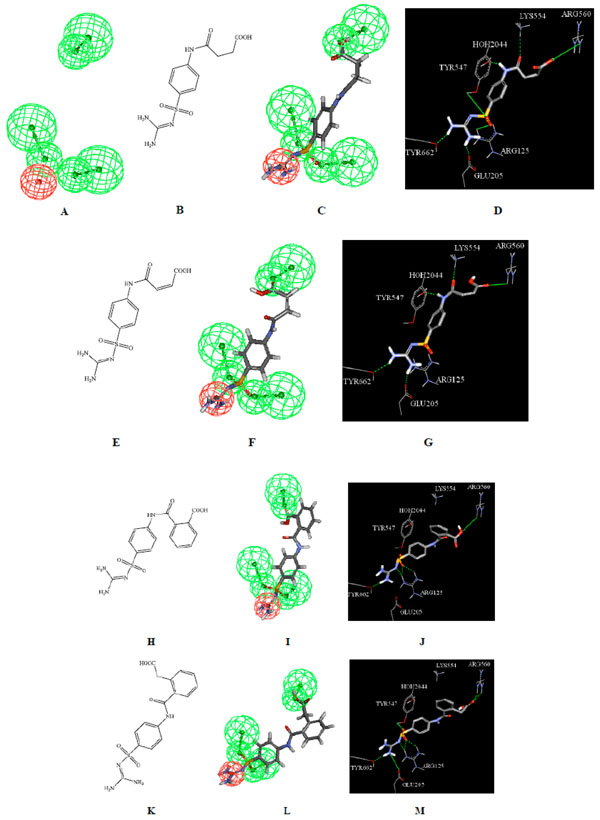
The mapping of Hypo4/10 against the synthesized compounds 10, 11, 16 and 21. A) Hypo4/10 (hydrogen bond acceptors are represented as green vectored spheres, positive ionizable groups as red spheres) [17], B) structure of 10, C) Hypo4/10 mapped against 10, D) docking of 10 in DPP IV binding pocket (PDB 2JID), E) structure of 11, F) Hypo4/10 mapped against 11, G) docking of 11 in DPP IV binding pocket, H) structure of 16, I) Hypo4/10 mapped against 16, J) docking of 16 in DPP IV binding pocket, K) structure of 21, L) Hypo4/10 mapped against 21, M) docking of 21 in DPP IV binding pocket.
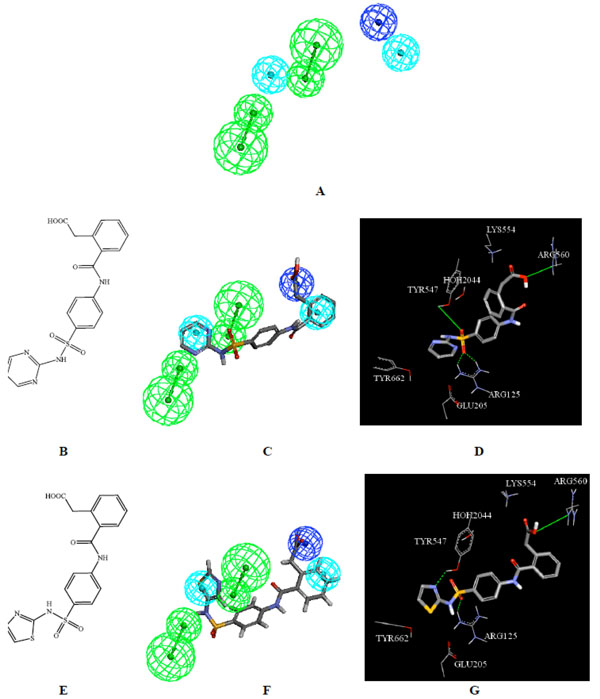
The mapping of Hypo32/8 against the synthesized compounds 17 and 18. A) Hypo32/8 (hydrogen bond acceptors are represented as green vectored spheres, negative ionizable groups as dark blue spheres, and hydrophobic features as blue spheres) [17], B) structure of 17, C) Hypo32/8 mapped against 17, D) Docking of 17 in DPP IV binding pocket (PDB: 2JID), E) structure of 18, F) Hypo32/8 mapped against 18, G) Docking of 18 in DPP IV binding pocket (PDB: 2JID).
Guided by the previous modeling efforts; sulfonamides were used as a basic scaffold for the design and synthesis of a series of N4-sulfonamido-succinamic, phthalamic, acrylic and benzoyl acetic acid derivatives (6-21, Scheme 1). The synthesized compounds were evaluated for their in vitro anti-DPP IV activity using commercially available DPP IV inhibitor screening assay kit (Table 1).
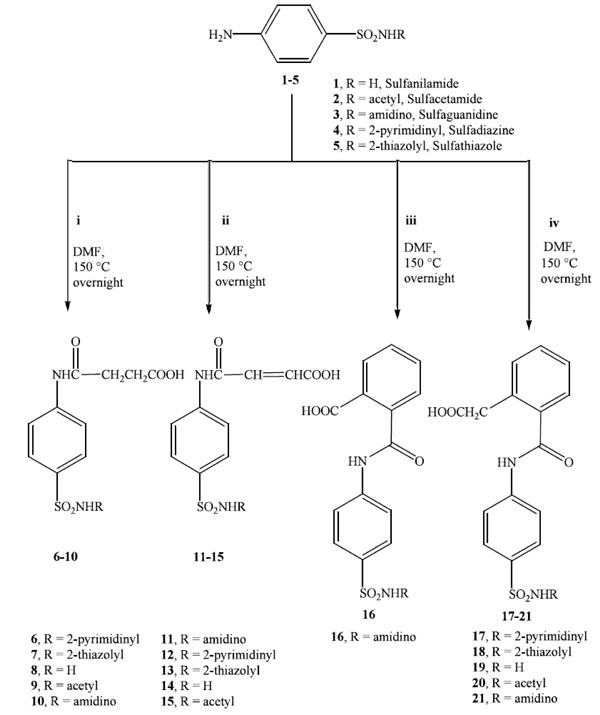
Synthesis of N4-sulfonamido-succinamic, phthalamic, acrylic and benzoyl acetic acid derivatives (6-21). i: succinic anhydride, ii: maleic anhydride, iii: phthalic anhydride and iv: homophthalic anhydride.
The synthesized N4-sulfonamido-succinamic, phthalamic, acrylic and benzoyl acetic acid derivatives 6-21 with their fit values against Hypo32/8 and Hypo4/10, their QSAR-Estimated IC50 and in vitro DPP IV bioactivities
| Compound | Fit values against Hypo32/8 a | Fit values against Hypo4/10 a | QSAR estimated IC50 (µM) b | In vitro DPP IV % inhibition c | In vitro IC50 (µM) e |
|---|---|---|---|---|---|
| 6 | 0 d | 0 | 0.5535 | 3 ± 0.2 | - |
| 7 | 0 | 0 | 0.0678 | 10 ± 0.5 | - |
| 8 | 0 | 0 | 0.0069 | 5 ± 0.3 | - |
| 9 | 0 | 0 | 0.1741 | 7 ± 0.7 | - |
| 10 | 0 | 7.3231 | 0.0028 | 23 ± 0.8 | 54.4 |
| 11 | 0 | 7.2859 | 0.0002 | 23 ± 0.3 | 43.2 |
| 12 | 0 | 0 | 0.0281 | 22 ± 0.9 | 54.6 |
| 13 | 0 | 0 | 0.0032 | 19 ± 1.5 | - |
| 14 | 0 | 0 | 0.0002 | 15 ± 1.0 | - |
| 15 | 0 | 0 | 0.0083 | 22 ± 1.1 | 59.3 |
| 16 | 0 | 7.2859 | 0.0022 | 15 ± 1.1 | - |
| 17 | 4.7265 | 0 | 20.200 | 25 ± 1.3 | 33.5 |
| 18 | 4.7265 | 0 | 3.8253 | 17 ± 0.5 | - |
| 19 | 0 | 0 | 0.6360 | 22 ± 0.9 | 48.5 |
| 20 | 0 | 0 | 18.683 | 15 ± 1.1 | - |
| 21 | 0 | 7.3076 | 0.6389 | 13 ± 0.4 | - |
| Gemifloxacin | 0 | 6.1005 | 0.0089 | 71 ± 1.0 | 2.7 |
a Best-fit values against each binding hypothesis calculated by the equation [23]:
Fit = ∑ mapped hypothesis features x W [1-∑ (disp/tol) 2]
b As calculated by QSAR equation (1) [17].
c Percentage inhibition at 10 µM concentration.
d Fit value of zero, means that the compound does not fit the hypothesis (misses a pharmacophoric feature).
e IC50 values were determined from the corresponding dose-response lines at three concentrations with a correlation coefficient of 0.99.
MATERIALS AND METHODS
General Methods
The proposed structures for compounds 6-21 were confirmed via IR spectroscopy, mass spectroscopy, 1H- and 13C-NMR spectra.
Melting points were measured using Gallenkampf melting point apparatus and are uncorrected. 1H NMR and 13C NMR spectra were collected on a Varian Oxford NMR300 and BRUKER NMR500 spectrometers. The samples were dissolved in deuterated DMSO. Mass spectrometry was performed using LC Mass Bruker Apex-IV mass spectrometer utilizing an electrospray interface.
Infrared spectra were recorded using Shimadzu IR Affinity-1 spectrophotometer. The samples were dissolved in CHCl3 and analysed for IR as thin solid films using NaCl plates. Analytical thin layer chromatography (TLC) was carried out using pre-coated aluminum plates and visualized by UV light (at 254 and/ or 360 nm). Column chromatography was carried out using high- purity grade silica (pore size 60A°, 70-230 mesh, 63-200 µm, Fluka).
Chemicals and solvents were purchased from corresponding companies (Sigma-Aldrich, Riedel-de Haen, Fluka, BDH Laboratory Supplies and Promega Corporation) and were used in the experimentation without further purification.
General procedure for synthesis of N4-sulfonamido-succinamic, phthalamic, acrylic and benzoyl acetic acid derivatives (6-21)
1 mmole of the benzenesulfonamide derivative 1-5 was dissolved in DMF (15 mL). Subsequently, 1.2 mmole of the required anhydride i-iv (succinic, maleic, phthalic and homophthalic, respectively) was added. The reaction mixture was left, under magnetic stirring, overnight at 150 °C. Afterward, the residue, after evaporation of the solvent, was purified either by recrystallization using CHCl3/MeOH (or CHCl3/EtOH) or by column chromatography eluting with CHCl3/MeOH (95:5) to give the desired N4-sulfonamido-succinamic, phthalamic, acrylic or benzoyl acetic acid derivative 6-21.
N-[4-l]-succinamic acid (6)
Off-white powder (55.0%): Rf = 0.72 (CHCl3-MeOH, 7:3); M.p. 230-231°C; IR (thin film) cm-1 3500, 3379, 3291, 3055, 2940, 1709, 1679, 1593, 1578, 1535, 1447, 1150; 1H-NMR (300 MHz, DMSO) δ 2.46 (t, J = 7.0 Hz, 2H), 2.52 (t, J = 7.0 Hz, 2H), 6.70 (t, J = 10 Hz, 1H), 7.60 (d, J = 15 Hz, 2H), 7.75 (d, J = 15 Hz, 2H), 8.27 (d, J = 10 Hz, 2H), 10.29 (s, 1H), 11.37-12.96 ppm (br s, 2H); 13C-NMR (300 MHz, DMSO) δ 29.5 (1C), 31.8 (1C), 113.4 (1C), 118.3 (2C), 128.8 (2C), 137.5 (1C), 142.3 (1C), 158.3 (2C), 160.7 (1C), 171.3 (1C), 174.5 ppm (1C); MS (ESI, positive mode) m/z [M+H]+ 351.05694 (C14H15N4O5S requires 351.35092).
N -[4-l]-succinamic acid (7)
Off-white powder (73.7%): Rf = 0.7 (CHCl3-MeOH, 7:3); M.p. 173-174°C; IR (thin film) cm-1 3507, 3329, 3285, 3102, 2901, 1713, 1674, 1593, 1570, 1497, 1146; 1H-NMR (300 MHz, DMSO) δ 2.04 (t, J = 7.0 Hz, 2H), 2.36 (t, J = 7.0 Hz, 2H), 6.77 (d, J = 10 Hz, 1H), 7.20 (d, J = 10 Hz, 1H), 7.68-7.71 (m, 4H), 10.28 (s, 1H), 12.61-13.13 ppm (br s, 2H); 13C-NMR (300 MHz, DMSO) δ 29.1 (1C), 31.5 (1C), 108.6 (1C), 118.9 (2C), 125.1 (1C), 127.5 (2C), 136.5 (1C), 143.9 (1C), 169.1 (1C), 171.3 (1C), 174.3 ppm (1C); MS (ESI, positive mode) m/z [M+H]+ 356.01810 (C13H14N3O5S2 requires 356.39154).
N-(4-Sulfamoyl-phenyl)-succinamic acid (8)
White powder (79.7%): Rf = 0.63 (CHCl3-MeOH, 7:3) ; M.p. 202-203°C; IR (thin film) cm-1 3530, 3350, 3213, 3113, 2943, 1715, 1673, 1597, 1543, 1130, 1147; 1H-NMR (300 MHz, DMSO) δ 2.24 (t, J = 6.9 Hz, 2H), 2.46 (t, J = 6.9 Hz, 2H), 7.21 (s, 2H), 7.43-7.70 (m, 4H), 10.29 (s, 1H), 11.95 ppm (br s, 1H); 13C-NMR (300 MHz, DMSO) δ 29.1 (1C), 31.6 (1C), 118.9 (2C), 127.2 (2C), 138.5 (1C), 142.7 (1C), 171.3 (1C), 174.4 ppm (1C); MS (ESI, positive mode) m/z [M+H]+ 273.03527 (C10H13N2O5S requires 273.27876).
N-(4-Acetylsulfamoyl-phenyl)-succinamic acid (9)
White powder (45.0%): Rf = 0.66 (CHCl3-MeOH, 7:3); M.p. 230-231°C; IR (thin film) cm-1 3504, 3271, 3183, 3125, 2913, 1720, 1672, 1593, 1543, 1470, 1150; 1H-NMR (500 MHz, DMSO) δ 1.91 (s, 3H), 2.54 (t, J = 7.0 Hz, 2H), 2.62 (t, J = 7.0 Hz, 2H), 7.78 (d, J = 8.9 Hz, 2H), 7.84 (d, J = 8.9 Hz, 2H), 10.45 (s, 1H), 11.88-12.25 ppm (br s, 2H); 13C-NMR (500 MHz, DMSO) δ 23.7 (1C), 29.1 (1C), 31.7 (1C), 118.8 (2C), 129.3 (2C), 133.2 (1C), 144.2 (1C), 169.1 (1C), 171.4 (1C), 174.2 ppm (1C); MS (ESI, positive mode) m/z [M+H]+ 316.04585 (C12H15N2O6S requires 316.31544).
N-[4-l]-succinamic acid (10)
White powder (77.4%): Rf = 0.81 (CHCl3-MeOH, 6:4); M.p. 218-219°C; IR (thin film) cm-1 3503, 3449, 3337, 3198, 3140, 2932, 1713, 1670, 1597, 1543, 1254; 1H-NMR (500 MHz, DMSO) δ 2.58 (t, J = 6.9 Hz, 2H), 2.64 (t, J = 6.9 Hz, 2H), 6.66 (br s, 4H), 7.68 (s, 4H), 10.24 (s, 1H), 12.14 ppm (br s, 1H); 13C-NMR (500 MHz, DMSO) δ 29.2 (1C), 31.6 (1C), 118.7 (2C), 127.0 (2C), 139.0 (1C), 142.1 (1C), 158.5 (1C), 171.1 (1C), 174.2 ppm (1C); MS (ESI, positive mode) m/z [M+H]+ 315.05973 (C11H15N4O5S requires 315.31882).
3-[4-l]-acrylic acid (11)
Yellow powder (47.3%): Rf = 0.60 (CHCl3-MeOH, 6:4); M.p. 180-181°C; IR (thin film) cm-1 3500, 3456, 3352, 3213, 3098, 2889, 1728, 1632, 1585, 1539, 1501, 1138; 1H-NMR (500 MHz, DMSO) δ 6.33 (d, J = 12 Hz, 1H), 6.49 (d, J = 12 Hz, 1H), 6.68 (br s, 4H), 7.70 (s, 4H), 10.59 (s, 1H), 12.91 ppm (br s, 1H); 13C-NMR (500 MHz, DMSO) δ 119.3 (2C), 127.1 (2C), 130.8 (1C), 132.0 (1C), 139.8 (1C), 141.5 (1C), 158.5 (1C), 164.0 (1C), 167.4 ppm (1C); MS (ESI, positive mode) m/z [M+H]+ 313.04064 (C11H13N4O5S requires 313.30294).
3-[4-l]-acrylic acid (12)
Off-white powder (83.8%): Rf = 0.76 (CHCl3-MeOH, 6:4); M.p. 210-211°C; IR (thin film) cm-1 3568, 3337, 3217, 3140, 3040, 2943, 1724, 1632, 1585, 1558, 1531, 1501, 1493, 1439, 1165; 1H-NMR (500 MHz, DMSO) δ 6.33 (d, J = 12 Hz, 1H), 6.4 (d, J = 12 Hz, 1H), 7.05 (t, J = 4.9 Hz, 1H), 7.79 (d, J = 8.8 Hz, 2H), 7.95 (d, J = 8.8 Hz, 2H), 8.51 (d, J = 4.9 Hz, 2H), 10.67 (s, 1H), 11.25-12.88 ppm (br s, 2H); 13C-NMR (500 MHz, DMSO) δ 116.3 (1C), 119.2 (2C), 129.4 (2C), 130.6 (1C), 132.2 (1C), 135.0 (1C), 143.1 (1C), 157.4 (1C), 158.8 (1C), 164.3 (1C), 167.3 ppm (2C); MS (ESI, positive mode) m/z [M+H]+ 349.03987 (C14H13N4O5S requires 349.33504).
3-[4-l]-acrylic acid (13)
Off-white powder (48.1%): Rf = 0.69 (CHCl3-MeOH, 6:4); M.p. 211-212°C; IR (thin film) cm-1 3504, 3337, 3225, 3136, 3013, 2893, 1724, 1632, 1582, 1543, 1497, 1153; 1H-NMR (500 MHz, DMSO) δ 6.33 (d, J = 12 Hz, 1H), 6.48 (d, J = 12 Hz, 1H), 6.82 (d, J = 6.9 Hz, 1H), 7.25 (d, J = 6.9 Hz, 1H), 10.63 (s, 1H), 12.52-13.06 ppm (br s, 2H); 13C-NMR (500 MHz, DMSO) δ 108.6 (1C), 119.4 (2C), 124.9 (1C), 127.4 (2C), 130.8 (1C), 132.0 (1C), 137.3 (1C), 142.3 (1C), 164.1 (1C), 167.4 (1C), 169.2 ppm (1C); MS (ESI, positive mode) m/z [M+H]+ 354.01859 (C13H12N3O5S2 requires 354.37566).
3-(4-Sulfamoyl-phenylcarbamoyl)-acrylic acid (14)
White powder (75.5%): Rf = 0.69 (CHCl3-MeOH, 6:4); M.p. 190-191°C; IR (thin film) cm-1 3500, 3375, 3271, 3121, 3067, 2855, 1713, 1655, 1589, 1539, 1443, 1242; 1H-NMR (500 MHz, DMSO) δ 6.34 (d, J = 12 Hz, 1H), 6.50 (d, J = 12 Hz, 1H), 7.27 (s, 2H), 7.78 (s, 4H), 10.63 (s, 1H), 12.93 ppm (br s, 1H); 13C-NMR (500 MHz, DMSO) δ 119.4 (2C), 127.2 (2C), 130.8 (1C), 131.9 (1C), 139.2 (1C), 142.1 (1C), 164.1 (1C), 167.4 ppm (1C); MS (ESI, positive mode) m/z [M+H]+ 271.01940 (C10H11N2O5S requires 271.26288).
3-(4-Acetylsulfamoyl-phenylcarbamoyl)-acrylic acid (15)
Off-white powder (41.2%): Rf = 0.73 (CHCl3-MeOH, 6:4); M.p. 195-196°C; IR (thin film) cm-1 3503, 3294, 3201, 3113, 3075, 2878, 1710, 1690, 1628, 1589, 1551, 1466, 1165; 1H-NMR (500 MHz, DMSO) δ 1.92 (s, 3H), 6.34 (d, J = 12 Hz, 1H), 6.51 (d, J = 12 Hz, 1H), 7.83 (d, J = 8.8 Hz, 2H), 7.88 (d, J = 8.8 Hz, 2H), 10.73 (s, 1H), 12.00 (br s, 1H), 12.89 ppm (br s, 1H); 13C-NMR (500 MHz, DMSO) δ 23.7 (1C), 119.4 (2C), 129.4 (2C), 130.6 (1C), 132.2 (1C), 133.8 (1C), 143.7 (1C), 162.8 (1C), 167.3 (1C), 169.2 ppm (1C); MS (ESI, positive mode) m/z [M+H]+ 313.02977 (C12H13N2O6S requires 313.29956).
N-[4-l]-phthalamic acid (16)
White powder (33.8%): Rf = 0.81 (CHCl3-MeOH, 6:4); M.p. 243-244°C; IR (thin film) cm-1 3506, 3445, 3341, 3233, 3156, 1717, 1628, 1597, 1535, 1504, 1466, 1261; 1H-NMR (500 MHz, DMSO) δ 6.54 (d, J = 8.6 Hz, 1H), 6.78 (br s, 4H), 7.39 (d, J = 8.6 Hz, 1H), 7.65 (d, J = 8.5 Hz, 2H), 7.71 (m, 1H), 7.91 (d, J = 8.5 Hz, 2H), 8.01 (m, 1H), 10.30 (s, 1H), 12.03 ppm (br s, 1H); 13C-NMR (500 MHz, DMSO) δ 112.8 (1C), 124.0 (2C), 126.7 (2C), 127.2 (2C), 130.7 (1C), 134.7 (1C), 135.3 (2C), 144.3 (1C), 158.2 (1C), 158.7 (1C), 167.2 ppm (1C); MS (ESI, positive mode) m/z [M+H]+ 363.06465 (C15H15N4O5S requires 363.36162).
{2-[4-l]-phenyl}-acetic acid (17)
Off-white powder (64.6%): Rf = 0.90 (CHCl3-MeOH, 85:15); M.p. 233-234°C; IR (thin film) cm-1 3588, 3372, 3215, 3105, 2866, 1697, 1678, 1589, 1493, 1443, 1161; 1H-NMR (500 MHz, DMSO) δ 4.12 (s, 2H), 7.04 (t, J = 10 Hz, 1H), 7.37 (dd, J = 10, 5.0 Hz, 2H), 7.52 (t, J = 10 Hz, 1H), 7.74 (dd, J = 10, 5.0 Hz, 2H), 7.91 (t, J = 10 Hz, 3H), 8.50 (d, J = 10 Hz, 2H), 10.48 (s, 1H), 11.57-12.93 ppm (br s, 2H); 13C-NMR (500 MHz, DMSO) δ 42.6 (1C), 116.3 (1C), 118.6 (2C), 127.5 (1C), 129.3 (2C), 130.8 (1C), 131.3 (1C), 132.2 (1C), 132.9 (1C), 134.1 (1C), 136.9 (1C), 143.9 (1C), 157.4 (2C), 158.8 (1C), 168.9 (1C), 170.4 ppm (1C); MS (ESI, positive mode) m/z [M+H]+ 413.46201 (C19H17N4O5S requires 413.42030).
{2-[4-l]-phenyl}-acetic acid (18)
Off-white powder (73.9%): Rf = 0.85(CHCl3-MeOH, 85:15); M.p. 231-232°C; IR (thin film) cm-1 3566, 3399, 3152, 3105, 2913, 1715, 1660, 1589, 1574, 1535, 1146; 1H-NMR (500 MHz, DMSO) δ 4.12 (s, 2H), 6.80 (d, J = 10 Hz, 1H), 7.23 (d, J = 10 Hz, 1H), 7.36-7.40 (m, 2H), 7.50-7.54 (m, 1H), 7.70-7.75 (m, 4H), 7.80-7.91 (m, 1H), 10.38 (s, 1H), 12.55-12.86 ppm (br s, 2H); 13C-NMR (500 MHz, DMSO) δ 42.5 (1C), 108.5 (1C), 118.9 (2C), 124.9 (1C), 127.4 (2C), 130.7 (1C), 131.4 (1C), 132.2 (1C), 132.8 (1C), 136.5 (1C), 137.0 (1C), 143.1 (1C), 168.9 (2C), 169.1 (1C), 170.2 ppm (1C); MS (ESI, positive mode) m/z [M+H]+ 418.03642 (C18H16N3O5S2 requires 418.46092).
[2-l]-acetic acid (19)
Off-white powder (62.9%): Rf = 0.80(CHCl3-MeOH, 85:15); M.p. 233-234°C; IR (thin film) cm-1 3560, 3348, 3318, 3256, 3102, 2870, 1694, 1651, 1593, 1574, 1520, 1497, 1157; 1H-NMR (500 MHz, DMSO) δ 4.13 (s, 2H), 7.23 (s, 2H), 7.37-7.41 (m, 2H), 7.51-7.55 (m, 1H), 7.72-7.76 (m, 4H), 7.90-7.92 (m, 1H), 10.42 (s, 1H), 12.84 ppm (br s, 1H); 13C-NMR (500 MHz, DMSO) δ 42.5 (1C), 118.8 (2C), 127.1 (2C), 127.4 (1C), 130.8 (1C), 131.4 (1C), 132.2 (1C), 132.9 (1C), 137.0 (1C), 138.4 (1C), 142.9 (1C), 169.0 (1C), 170.2 ppm (1C); MS (ESI, positive mode) m/z [M+H]+ 335.05429 (C15H15N2O5S requires 335.34814).
[2-l]-acetic acid (20)
White powder (41.4%): Rf = 0.88 (CHCl3-MeOH, 85:15); M.p. 218-219°C; IR (thin film) cm-1 3550, 3329, 3294, 3032, 2870, 1721, 1694, 1667, 1593, 1528, 1443, 1165; 1H-NMR (500 MHz, DMSO) δ 1.92 (s, 3H), 4.14 (s, 2H), 7.37-7.41 (m, 2H), 7.51-7.55 (m, 1H), 7.78 (d, J = 10 Hz, 2H), 7.84 (d, J = 10 Hz, 2H), 7.90-7.92 (m, 1H), 10.50 (s, 1H), 11.95 (br s, 1H), 12.76 ppm (br s, 1H); 13C-NMR (500 MHz, DMSO) δ 23.7 (1C), 42.6 (1C), 118.8 (2C), 127.5 (1C), 129.3 (2C), 130.8 (1C), 131.3 (1C), 132.2 (1C), 132.9 (1C), 133.1 (1C), 136.9 (1C), 144.5 (1C), 168.9 (1C), 169.1 (1C), 170.5 ppm (1C); MS (ESI, positive mode) m/z [M+H]+ 377.06537 (C17H17N2O6S requires 377.38482).
{2-[4-l]-phenyl}-acetic acid (21)
White powder (66.7%): Rf = 0.60 (CHCl3-MeOH, 85:15); M.p. 223-224°C; IR (thin film) cm-1 3570, 3456, 3426, 3333, 3194, 1694, 1678, 1597, 1520, 1404, 1138; 1H-NMR (500 MHz, DMSO) δ 4.12 (s, 2H), 6.64 (br s, 4H), 7.37-7.41 (m, 2H), 7.51-7.54 (m, 1H), 7.67 (s, 4H), 7.89-7.91 (m, 1H), 10.32 (s, 1H), 12.80 ppm (br s, 1H); 13C-NMR (500 MHz, DMSO) δ 42.4 (1C), 118.7 (2C), 127.0 (2C), 127.4 (1C), 130.7 (1C), 131.4 (1C), 132.2 (1C), 132.8 (1C), 137.0 (1C), 139.0 (1C), 142.3 (1C), 158.5 (1C), 168.9 (1C), 170.1 ppm (1C); MS (ESI, positive mode) m/z [M+H]+ 377.07629 (C16H17N4O5S requires 377.38820).
DPP IV Inhibition Assay
DPP IV inhibitory bioactivities were assayed using commercially available kit (Cayman's DPP IV Inhibitor
Screening Assay Kit, USA) which provides a fluorescence-based method for screening DPP IV inhibitors. The assay uses the fluorogenic substrate, Gly-Pro-Amino-methylcoumarin (AMC), to measure DPP IV activity. Cleavage of the peptide bond by DPP IV releases the free AMC group, resulting in fluorescence that is measured at an excitation wavelength of 350-360 nm and an emission wavelength of 450-465 nm. Inhibition of DPP will prevent AMC release and therefore decrease fluorescence intensity.
The tested compounds were initially dissolved in DMSO to yield 10 mM stock solutions and subsequently diluted to the required concentrations using distilled deionized water and then were added to the assay well in a final volume of 10 μl (and a final concentration of 10 μM). The final concentration of DMSO was adjusted to 0.1 %. The assay procedure can be described briefly as follows: 30 μl of assay buffer, 10 μl of diluted DPP, and 10 μl of the inhibitor were added to the inhibitor wells. The reaction was initiated by adding 50 μl of diluted Substrate Solution. The plate was covered and incubated for 30 minutes at 37 °C. After incubation, fluorescence intensity (Excitation λ:350 nm; Emission λ:465 nm) was read in a FLX800TBI Microplate Fluorimeter (BioTek Instruments, Winooski, USA). The assay is carried out using a black 96-well plate with led.
The percentage of residual activity of DPP IV was determined for each compound by comparing the activity of DPP IV in the presence and absence of the tested compound. DPP IV activity was not affected at the used DMSO concentration. Negative controls lacking DPP IV were used as background. A standard DPP IV inhibitor, Gemifloxacin, was used as a positive control. All measurements were conducted in duplicates.
Docking and Scoring
The binding site of DPP IV was generated from the co-crystallized ligand GVB1771 within the targeted protein (PDB code: 2JID, resolution 2.8 A°). The ligands were docked using LigandFit (Accelrys DS Studio 2.1) and default parameters [29,30]. The resulting docked poses were scored employing PLP1 [31], PLP2 [32], ligscore1 [29], ligscore 2 [29], PMF [33,34] and JAIN [35-37].
RESULTS AND DISCUSSION
Chemistry
In the current work, the desired N4-sulfonamido-succinamic, phthalamic, acrylic and benzoyl acetic acid derivatives 6-21 were prepared from the reaction of one of the benzenesulfonamide derivatives 1-5 with the required anhydride i-iv (succinic, maleic, phthalic and homophthalic, respectively) in dimethylformamide at 150 °C with overnight reflux, as illustrated in Scheme 1. Sulfonamides 6-16 were synthesized earlier by other research groups [38-41]. While compounds 17-21 are novel sulfonamide derivatives. The highest yield was obtained upon reacting 4-amino-N-pyrimidin-2-yl-benzenesulfonamide 4 with maleic anhydride ii to give 12 in 83.8% yield.
Scheme 1 shows the synthesized N4-sulfonamido-succinamic, phthalamic, acrylic and benzoyl acetic acid derivatives (6-21).
The synthesized compounds were designed to enclose the desired pharmacophoric features responsible for binding to DPP IV receptor site [17]. The chemical features considered were hydrogen bond acceptors, positive ionizable groups, negative ionizable groups and hydrophobic features (Fig. 2 and Fig. 3).
Biological Evaluation
The results of DPP IV inhibition assay, presented in Table 1, demonstrate that compound 17 exhibit appreciable activity against DPP IV. Although our newly synthesized compounds are of lower potency than some published DPP IV inhibitors, these derivatives are characterized by their novel scaffold that can serve as a promising lead for further optimization. Table 1 shows the fit values of the synthesized compounds against Hypo32/8 and Hypo4/10, together with their QSAR-estimated IC50 values and their in vitro bioactivities.
By comparing the synthesized N4-sulfonamido-succinamic acid derivatives 6-10 (Scheme 1, Table 1), it was found that compound 10 has the highest activity with 23% inhibition at 10 μM concentration. This can be attributed to the presence of the amidino moiety in compound 10 that is mapped as a positive ionizable feature by Hypo4/10.
Similar results were found when evaluating the synthesized N4-sulfonamido-acrylic acid derivatives 11-15 (Scheme 1, Table 1), where compound 11 enclosing the amidino group showed the optimum inhibition. Compound 12 also showed similar inhibition effect as the amidino derivatives which could be attributed to sharing equivalent interactions within the DPP IV binding pocket since similar pharmacophoric features lead to similar activities.
In contrast, regarding the synthesized N4-sulfonamido- benzoyl acetic acid derivatives 17-21 (Scheme 1, Table 1), compound 17 had the highest activity with 25% inhibition (IC50= 33.5 μM). This can be caused by the existence of the pyrimidine ring in 17 that is mapped as both hydrophobic and hydrogen bond acceptor feature by Hypo32/8.
Fig. (2) shows how Hypo4/10 maps the synthesized compounds 10, 11, 16 and 21. The positive ionizable feature of Hypo4/10 fits the diaminomethylene group of these compounds, while one of the hydrogen bond acceptor features maps the carboxylic acid group. Furthermore, the other two hydrogen bond acceptors of Hypo4/10 fit the sulfone moiety in the structures.
On the other hand, Fig. (3) shows how Hypo32/8 fits the synthesized compounds 17 and 18. The negative ionizable feature of Hypo32/8 maps the carboxylic acid group of 17 and 18, while the two hydrophobic features fit the lateral aromatic ring and the heterocycle i.e. pyrimidine ring of 17 and thiazole ring of 18. Additionally, the two hydrogen bond acceptors of Hypo32/8 map the sulfone group and the nitrogen atom of the heterocycle.
As a general trend, the activity of the synthesized sulfonamido-derivatives was shown to be affected by the chemical properties of substituents on both N1 and N4. The presence of basic N1 substituent, which is in accordance with the positive ionizable feature of Hypo4/10, together with a rigid acidic N4 group, may contribute to the increase in DPP IV inhibitory bioactivity. Furthermore, the presence of an electron withdrawing group capable of accepting hydrogen bond may also enhance the inhibitory bioactivity.
Additionally, the hydrophobic aromatic ring present at N4 may also enhance the inhibitory activity as can be seen in molecules 16-21. Moreover, the presence of carboxylic acid group and a hydrophobic aromatic ring agrees well with the pharmacophoric features of Hypo32/8.
The pharmacophore features obtained by structure-based pharmacophore modeling can be compared with the structure of DPP IV binding site to identify probable residues important for inhibition. The docking parameters output is summarized in Table 2 (under Supplementary Material). Furthermore, illustrating figures that show the interaction of selected synthetic compounds 10, 11, 16, 17, 18 and 21 with the amino acids in the DPP IV binding pocket are shown in figures 2d, 2g, 2j, 2m, 3d and 3g. The figures show the interaction between the functional groups that correspond to the pharmacophoric features. A marked similarity was observed between the features proposed by the pharmacophore models and the ligand binding features in the docked structures. For example, negative ionizable feature in Hypo32/8 corresponds to the interaction with the guanidino group of Arg560, hydrophobic features correspond to stacking with aromatic rings of Tyr547 and Tyr662 while hydrogen bond acceptors correspond to OH group of Tyr547 and guanidino of Arg125 as shown in (Fig. 3).
Whereas Hypo4/10 contains three hydrogen bond acceptors that correspond to the interactions with NH2 group of Lys554, bridging Water HOH2044, OH group of Tyr662, guanidino group of Arg125, while positive ionizable feature corresponds to carboxylate group of Glu205 as shown in (Fig. 2).
It is important to realize the fact that not all amino acids in the binding pocket are essential for activity, only certain interactions with selected amino acids could be effective as inhibitor, this point was clarified using dbCICA technique [42].
To conclude, the presence of positive and negative ionizable groups combined with electron with drawing moieties that act as hydrogen bond acceptors, together with hydrophobic aromatic ring are found to be necessary features for in vitro DPP IV bioactivity.
Indeed, the synthesized N4-sulfonamido-succinamic, phthalamic, acrylic and benzoyl acetic acid compounds 6-21 were shown to have common pharmacophoric features with our DPP IV inhibitor lead, Gemifloxacin. Further research work and study to re-arrange and enhance better matching of these groups with the pharmacophoric features of Hypo4/10 and Hypo32/8 are required in order to improve the DPP IV inhibitory bioactivity.
CONCLUSION
In conclusion, we have successfully accomplished synthetic investigation of a series of N4-sulfonamido-succinamic, phthalamic, acrylic and benzoyl acetic acid derivatives as potential DPP IV inhibitors. Future optimization of these derivatives can lead to the discovery of new potent compounds.
CONFLICT OF INTEREST
None of the authors of the above manuscript has declared any conflict of interest which may arise from being named as an author on the manuscript.
SUPPLEMENTARY MATERIAL
Supplementary material can be viewed at Download Supplementary Material
ACKNOWLEDGEMENT
The authors acknowledge the Scientific Research and Postgraduate Deanship at Al-Zaytoonah University of Jordan for sponsoring this project. Authors thank the research assistant Ghadeer Albadawi for her great help during the synthesis of the targeted molecules.
REFERENCES
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link] [PMC Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]
[PubMed Link]

