All published articles of this journal are available on ScienceDirect.

Nitropropenyl Benzodioxole, An Anti-Infective Agent with Action as a Protein Tyrosine Phosphatase Inhibitor
Abstract
We report on the activities of a broad spectrum antimicrobial compound,nitropropenyl benzodioxole (NPBD) which are of relevance to its potential as an anti-infective drug. These investigations support the proposal that a major mechanism of NPBD is action as a tyrosine mimetic, competitively inhibiting bacterial and fungal protein tyrosine phosphatases (PTP).
NPBD did not affect major anti-bacterial drug targets, namely, ATP production, cell wall or cell membrane integrity, or transcription and translation of RNA. NPBD inhibited bacterial YopH and human PTP1B and not human CD45 in enzyme assays. NPBD inhibited PTP-associated bacterial virulence factors, namely, endospore formation in Bacillus cereus, prodigiosin secretion in Serratia marcescens, motility in Proteus spp., and adherence and invasion of mammalian cells by Yersinia enterocolitica. NPBD acts intracellularly to inhibit the early development stages of the Chlamydia trachomatis infection cycle in mammalian cells known to involve sequestration of host cell PTPs. NPBD thus both kills pathogens and inhibits virulence factors relevant to early infection, making it a suitable candidate for development as an anti-infective agent, particularly for pathogens that enter through, or cause infections at, mucosal surfaces. Though much is yet to be understood about bacterial PTPs, they are proposed as suitable anti-infective targets and have been linked to agents similar to NPBD. The structural and functional diversity and heterogeneous distribution of PTPs across microbial species make them suitably selective targets for the development of both broadly active and pathogen-specific drugs.
1. INTRODUCTION
Protection from infectious disease relies on the effective management of current antimicrobial agents, the development of new classes of compounds, and the identification of new microbial targets in order to address the serious problems caused by the emergence of drug-resistant human and animal pathogens [1]. Compounds currently in development predominantly address new targets in well-known target pathways. Several have a new target and mechanism of action, including action on membrane integrity and permeability and chelation of microbial metallo-enzymes [2, 3]. Unfortunately, developmental drugs have a low probability of reaching and completing clinical development [1, 4] so the need to identify new targets and agents remains perpetually urgent.
The antimicrobial properties of several benzyl nitroethenes have been reported [5-8]. Nicoletti et al. [5] have investigated the structure-activity relationships (SAR) of 23 benzyl nitroalkenes against a panel of clinically significant bacterial [9] and fungal species [7]. The SAR study showed the importance of the nitroethenyl and nitropropenyl side chain to anti-microbial activity, proposed by Park and Pei to be essential for inhibition of protein tyrosine phosphatases (PTP), with the nitropropenyl substituent being the most active [10]. The 23 compounds showed broad antimicrobial activity that differed across species, with greatest activity against Gram-positive bacteria and fungi and least against enteric Gram-negative rods. One of the most active compounds, nitropropenyl benzodioxole (NPBD) (Fig. 1) a tyrosine mimetic, is being investigated for development as an anti-infective agent. It is broadly microbicidal to many Gram-negative and most Gram-positive bacterial species and to all fungal species tested to date, and is active against strict anaerobes and intracellular pathogens including Mycobacterium tuberculosis and Plasmodium falciparum [5, 11, 12]. It has low oral toxicity for mice and chickens and is poorly absorbed from the intestinal tract [5, 13]. It also fails to be induced or selected for resistant strains of major bacterial and yeast pathogens after 16 week exposure in vitro [11].
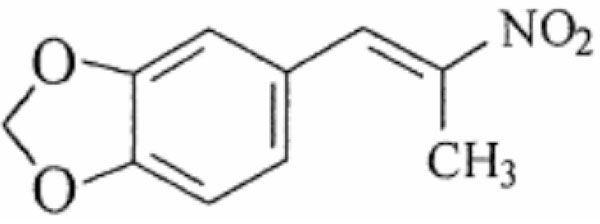
Structure of 5-[(E)-2-nitroprop-1-enyl]-1,3-benzodioxole (NPBD).
This paper reports investigations into the possible mechanisms of action of NPBD as a growth inhibitor in bacteria, including action on the major targets of existing anti-infective drugs. Given NPBD’s characteristic as a tyrosine mimetic capable of inhibiting protein tyrosine phosphatases (PTPs), bacterial processes known to involve PTPs and which are significant in virulence and early stages of infection are presented. These studies indirectly confirm PTP inhibition as a major mechanism of action and are relevant to its suitability for development as an anti-infective drug.
Phosphorylation and dephosphorylation of tyrosine, serine and threonine residues are major mechanisms for post-translational modification of proteins that control intracellular signalling in eukaryotes and prokaryotes [14-16]. Of the protein-bound phosphate in higher eukaryotes, 95% is on serine, ~5% on threonine, and 0.01 to 0.1% on tyrosine [17]. Protein tyrosine phosphatases (PTPs) belong to a large and structurally diverse family which share a common highly conserved catalytic site, with an invariant PTP signature motif that forms the tyrosine phosphate-binding loop [15, 18]. PTPs can be ligand-controlled transmembrane proteins or cytoplasmic. Regulatory sequences flanking the catalytic site in non-transmembrane PTPs influence sub-cellular distribution, binding and substrate selectivity [14]. Such variations may allow for selective inhibition of PTPs involved in survival, pathogenesis or virulence of micro-organisms.
Eukaryotic PTPs regulate cellular activities such as the cell cycle, growth and differentiation, cell to cell communication, gene transcription, the immune response and apoptosis. PTPs and protein tyrosine kinases (PTK) have been reported in a variety of bacterial species [19, 20]. There are several classes of bacterial PTPs including conventional, eukaryotic-like PTPs, low molecular weight PTPs and dual specific phosphatases (DSP) that dephosphorylate serine, threonine or tyrosine. Conventional PTPs are excreted and involved in the dephosphorylation of proteins of higher organisms during adhesion and invasion by bacterial cells [21]. PTPs in bacteria function differently from those in eukaryotes, showing greater catalytic versatility and a heterogeneous distribution across species. They play a role in aggression, survival and responses to environmental stimuli [19]. Many PTPs are virulence factors for pathogenic bacteria. The outer membrane proteins of Yersinia spp. (Yops) are inserted into host cells by type III secretion systems disrupting immune system signal cascades in host cells, enabling bacteria to survive and replicate [22]. Low molecular weight PTPs are involved in exopolysaccharide formation and biofilm formation in Pseudomonas spp. [23], exopolysaccharide secretion in Escherichia coli [9], capsule production in Streptococcus pneumoniae [24] and Klebsiella pneumonia [25] and flagellin export in Pseudomonas spp. [26]. Whitmore and Lamont [27] have reviewed the role of PTPs in bacterial virulence.
To combat increasing numbers of bacterial pathogens resistant to current drugs and the lack of anti-infective agents to treat infections with resistant phenotypes, compounds attacking virulence mechanisms have been investigated as potential therapeutic agents [28]. PTP inhibitors have been investigated for effect on toxins, biofilm formation, quorum sensing, cell adhesion and type III secretion systems [17, 29-31]. PTK inhibitors are successfully used in cancer therapy [32] and PTP inhibitors are potential oncology drugs [33]. Miller et al. [34] have investigated tyrosine kinase inhibitors as antibacterial compounds. However, few antibacterial PTP inhibitors have been reported [35-39].
The functional diversity and heterogeneous distribution of PTPs across genera and between species of the same genus suggest they could be attractive targets for the development of anti-infective agents. Dephosphorylation can activate or inhibit phosphate-dependent signalling. The effect of phosphatase inhibitors therefore may promote or impede cellular activities depending on the presence and function(s) of the regulating PTPs.
Anti-infective drugs may have more than one mechanism of action. PTPs are involved in cell growth and differentiation in eukaryotes and may play similar roles in prokaryotes. We therefore investigated the effect of NPBD, as the lead analog, on bacterial metabolic functions involved in cell growth that are major targets for anti-infective drugs: cell wall and cytoplasmic membrane integrity, membrane-bound ATP production and RNA transcription and translation. We report on its action on bacterial virulence mechanisms known to specifically involve PTP signalling: motility, pigment production, endospore formation and cell adherence and invasion mechanisms. As obligate intracellular parasites with restricted metabolic capacity, Chlamydia spp. are a suitable cell model for investigating the effect of a PTP inhibitor on the chlamydial development cycle and the host cell. Tyrosine kinases and phosphatases, both bacterial and human, have been shown to be involved in chlamydial entry and replication [40-42]. The bacterial species investigated cover a range of clinically significant pathogens for which more is known of PTP functions.
2. MATERIALS AND METHODS
Chemicals and enzymes were purchased from Merck KGaA and Sigma-Aldrich Co. LLC. Tris and ethylenediaminetetraacetic acid (EDTA) were from Amresco LLC. Minimum inhibitory concentrations (MICs) were determined using standardised methods [43, 44]. Assay plates, 96 well, were from Greiner Bio-One. Bacterial culture media was from Oxoid Ltd. Brucella broth was from BD biosciences. Cell culture media and reagents were from Thermo Fisher Scientific Inc. Cell culture plates were from Corning. Reagents and consumables for TEM were from ProSciTech Pty Ltd. All broth cultures of bacteria were incubated aerobically at 37°C with orbital shaking at 160 rpm unless otherwise stated. Viable counts were performed using 10-fold dilutions of broth cultures incubated on NA unless otherwise stated. All agar plates were incubated aerobically at 37°C for 24 h unless otherwise stated.
2.1. Protein Tyrosine Phosphatase Activity
Phosphatase activity in the presence of NPBD was investigated using a Tyrosine Phosphatase Assay (ProFluor™, Promega). Tyrosine phosphatases PTP1B and Yop and the serine/threonine DSP, CD45, dephosphorylate bisamide rhodamine 110 phosphopeptide (R110), allowing cleavage by a protease and release of fluorescence directly proportional to phosphatase activity. A protease-degradable fluorogenic peptide linked to 7-amino-4-methyl coumarin (AMC) allows identification of protease inhibitors. Assays were performed in black, 96-well immunoassay plates at wavelengths suitable to distinguish signals from both R110 (excitation 485 nm, emission 520 nm) and AMC substrates (excitation 355 nm, emission 460 nm) (Polarstar Omega, BMG Laboratories). PTP1B, Yop, and CD45 had specific activities of 50 U/mg protein, 80,000 U/mg protein and 125,000 U/mg protein respectively. Titration determined the optimal amount of enzyme to achieve 80% maximum fluorescence was 12 mU PTP1B, 3 U CD45 and 0.4 U Yop per well. Sodium orthovanadate, a phosphate analog and a general and potent PTP and DSP inhibitor [45], was included as a positive control. NPBD or sodium orthovanadate (5 μL, 10× in 10% DMSO) were added to duplicate wells to give 4-fold test concentrations of 0.03 μM to 160 μM for NPBD and 0.003 µM to 16 µM for sodium orthovanadate. Controls received 5 μL 10% DMSO only. Assay wells received 20 µL phosphatase and negative reaction controls received 20 µL buffer. Reactions were initiated with 25 µL peptide solution. Plates were incubated for 60 min at room temperature before the addition of protease solution (25 µL). After further 30 minutes incubation at room temperature, fluorescence stabiliser solution (25 µL) was added and fluorescence recorded. To demonstrate specific phosphatase inhibition, data were required to meet the following criteria: AMC fluorescence coefficient of variation ≤ 25%; maximum R110 fluorescence >10,000 U; and positive enzyme control well R110 fluorescence ≥ 70%. Duplicated readings from two separate assays were averaged and phosphatase activity (% maximum R110 fluorescence) plotted against log10 inhibitor concentrations (μM) ±SEM for each enzyme.
2.2. Bacterial ATP Synthesis
The Bac-Titre™ Glo assay (Promega) for detection of total adenosine 5-triphosphate (ATP) was used to investigate whether NPBD had an effect on ATP production in bacteria by measuring the oxidation of beetle luciferin to oxyluciferin by firefly luciferase. Overnight cultures diluted to ~1 × 106 colony forming units/mL (CFU/mL) in 10 mL Mueller-Hinton Broth (MHB) received NPBD at 16 and 32 µg/mL for B. cereus and 8 and 32 µg/mL for S. aureus. The final concentration of the DMSO vehicle was 1% in test cultures and controls. ATP was extracted from log phase aerobic broth cultures of B. cereus and Staphylococcus aureus exposed to NPBD for 0, 1, 2 and 4 h at 37°C. Luminescence and viable cell counts were performed at each time point. For luminescence measurements, culture samples were mixed with Bac-Titre™ Glo reagent (100 µL each) and Luminescence Units (LU) recorded (TD-20/20, Turner Designs). A standard curve using 10-fold concentrations of ATP disodium salt trihydrate in MHB showed LU were proportional to ATP concentrations between 0.1 and 100 nM (R2=0.9689). The standard curve was used to convert LU to units of ATP (1 nM = LU × 0.016 + 4.34). Correlation of ATP synthesis with viable count plotted against time was tested by regression analysis (Excel Data analysis, Microsoft).
2.3. RNA Transcription and Translation
The ‘E. coli S30 Extract for Circular DNA’ (Promega) was used to measure cell free protein synthesis in the presence of NPBD. A control DNA template (pGEM® β-Gal) containing the β-galactosidase coding sequence downstream from an E. coli lac Z promoter, allows direct spectrophotometric measurement of synthesised β-galactosidase. Conversion of colourless ONPG (o-nitrophenyl-β-D-galactopyranoside) to yellow ONP (o-nitrophenyl) is proportional to the amount of protein synthesised. The assay was adapted from Ulvatne et al. (2004) and performed according to manufacturer’s recommendations. A 96-well white immunoassay plate was prepared with 15 μL volume doubling dilutions of NPBD (8-128 µg/mL) in 60% v/v DMSO. A vehicle control received 15 μL of 60% v/v DMSO. The reaction control for maximum protein synthesis (no NPBD) and negative control (no extract, no NPBD) received nuclease free water. Amino acids (2 μL) and S30 premix (8 μL) were added to all wells. E. coli S30 extract (5 μL) was added to treatment wells and the reaction control. Nuclease-free water (20 μL) was added to bring well volumes to 50 µL and the plate incubated at 37°C for 30 min. DNA (1 μL, 0.25 mg/mL pGEM® β-Gal) was added to all wells and incubation continued for 30 min before adding 150 μL of 15 mM ONPG. Absorbance of ONP (A420nm) was recorded every 10 min for 2 h (Cary®50, Varian). Wells were prepared in triplicate and assays repeated three times. Mean absorbance was plotted against time and the rate of ONP production, indicative of the relative rate of protein synthesis, calculated as ‘slope of curve with NPBD/slope of positive reaction curve (without NPBD)’.
2.4. Integrity of the Cell Wall and Cytoplasmic Membrane
The effect of NPBD on the integrity of the wall and cytoplasmic membrane was assessed by whole cell lysis of log phase cultures of Candida albicans and B. cereus, and by lysis of spheroplasts of E. coli and Moraxella catarrhalis.
2.4.1. Whole Cell Lysis Assay
C. albicans and B. cereus were inoculated into 5 mL of Sabouraud liquid medium and MHB respectively and incubated for 2 h. A clear, 96-well flat bottomed microtitre plate was prepared with 100 µL NPBD and 100 µL culture in media. NPBD final concentrations were 32, 64 and 128 µg/mL in 1% v/v DMSO and the final inoculum 1.5 (108 CFU/mL. Blank wells received no bacteria. Plates were incubated aerobically at 37°C and absorbance (A600nm) read spectrophotometrically (Cary™50, Varian) immediately after inoculation, at 15, 30, 45 min and 1, 2, 4, 8 h. Microbicidal activity was measured by viable counts at 0, 2, 4 and 8 h. Lytic activity produces a decrease in viable cells accompanied by a decrease in turbidity. A decrease in cell viability with a non-proportional loss in turbidity indicates a non-lytic attack on cells. C. albicans and B. cereus were selected for comparison with the transmission electron microscopy (TEM) ultrastructure study.
2.4.2. Lysis of Spheroplasts
Overnight cultures of E. coli (MIC 256 μg/mL) and M. catarrhalis (MIC 16 μg/mL) in 10 mL of MHB were diluted 1/200 in 20 mL fresh MHB and incubated at 37°C for 2 h. Log phase cultures were centrifuged (5 min, 2000 ×g, 4°C) and washed twice in cold buffer (50 mM Tris, pH 8, 20% w/v sucrose). Cells were resuspended in 5 mL of buffer and each divided into 2× 2.5 mL for preparation of whole cells or spheroplasts. Spheroplasts were prepared according to the method of Ganzle et al. [46] by incubation with 1 mL 0.6 mg/mL lysozyme, 20% w/v sucrose and 0.06 M EDTA for 30 min at 37°C. Spheroplasts were checked with a phase contrast microscope for spherical appearance indicating absence of cell envelope. Preparations consisted of >80% E. coli and ~70% M. catarrhalis intact spheroplasts. Spheroplasts were collected by centrifugation (5 min, 500 ×g) and resuspended in 3 mL buffer with 10 mM CaCl2. Whole cell suspensions received 1.5 mL buffer and were incubated for 30 min at 37°C. Resuspended spheroplasts and whole cells were adjusted to A570nm 0.2. Each was added (100 μL) to a clear, flat-bottomed microtitre plate in duplicate wells. NPBD, 10 μL (10×, 50% DMSO/Phosphate buffered saline pH 7.4) was added to test wells. Controls received 10 µL of 50% DMSO/PBS. Turbidity was monitored (A570nm) every 10 min for 1 h. Average percentage changes in intact spheroplasts and whole cells were calculated over four assays according to the formula, 100×(A570 nm at time X / A570 nm at time 0). A decrease in absorbance indicates lysis of spheroplasts or whole cells. A decrease in the absorbance of spheroplasts exposed to NPBD indicates increased cytoplasmic membrane permeability compared to that of whole cells where the OM may prevent penetration by NPBD.
2.4.3. Gram-Negative Cell Envelope Permeability Assay
NPN (1-N-phenyl naphthylamine) is a hydrophobic fluorescent probe normally excluded by the lipopolysaccharide (LPS) layer of the Gram-negative bacterial outer membrane (OM) but if the LPS is disrupted it can partition into the hydrophobic core of the OM. The method was adapted from Helander and Mattila-Sandholm [47]. Mid-log phase E. coli, grown at 37°C for 2 h in 5 mL MHB, were centrifuged (10 min, 3000 ×g), washed and resuspended in 5 mM HEPES pH 7.2 at A600nm0.5. Viable cell counts confirmed the final inoculum was ~5 × 106 CFU/well. Wells of a clear 96-well flat bottomed microtitre plate were prepared with 50 µL of NPBD (1 mg/mL, 1% DMSO/HEPES), EDTA or polymyxin B. Final concentrations were 250 µg/mL for NPBD and for positive controls, 250 mM EDTA and 250 µM polymyxin B. Negative uptake controls received 50 µL HEPES alone. NPN (50 µL, 40 µM) was added to half the wells containing test compounds and the remaining wells received 50 µL HEPES. Bacterial suspension (100 µL) was added to all treatment wells immediately before beginning fluorescence readings. Fluorescence was measured within 5 min of addition bacteria at excitation 350 nm, emission 450 nm (Wallac 1420 Victor3™, Perkin Elmer). The assay was repeated three times and results represented as relative fluorescence values (corrected for fluorescence in the absence of NPN) and NPN-uptake values (relative fluorescence values as a ratio of those for the buffer control). An increase in fluorescence values is indicative of OM permeability as the hydrophobic compound moves into the hydrophobic core of the OM.
2.4.4. In Vitro Activity Against Lipopolysaccharide-Deficient Campylobacter Jejuni
The minimum inhibitory concentration (MIC) and the minimum bactericidal concentration (MBC) for NPBD against C. jejuni HB93-13 wild type and a LOS-deficient mutant (WaaF-) were compared by conventional assay. All C. jejuni cultures and assays were incubated for 48 h at 42°C under microaerophilic conditions (CampyGen, Oxoid). C. jejuni cultures were grown on horse blood agar with Skirrow’s Campylobacter growth supplement and assay inocula prepared as 18 h cultures in Brucella broth with the supplement. Inoculum densities were confirmed by viable plate counts on horse blood agar. Assays were performed in sterile vials prepared with 1 mL doubling dilutions of 2× NPBD or Brucella broth alone and inoculated with 1 mL volumes of the C. jejuni test strains. Samples from vials showing no turbid growth at 48 h were plated onto horse blood agar to determine the minimum microbicidal concentration.
2.5. Ultra-Morphology by Transmission Electron Microscopy (TEM)
Ultrastructural changes in B. cereus and C.albicans exposed to sub-inhibitory concentrations of NPBD were investigated by TEM. B. cereus and C. albicans were grown overnight in MHB and Sabouraud liquid medium respectively and diluted to ~1 × 106 CFU/mL in 5 mL fresh medium. NPBD was added to 0.5, 1 and 2 µg/mL for B.cereus and 2, 4 and 8 µg/mL for C. albicans with 1% DMSO. Broth cultures were incubated for 24 h and viable counts performed. Reagent dilutions and washing were with 0.1 M sodium cacodylate buffer. Treated cultures (1.5 mL) were pelleted in 4°C at 1,180 ×g for 5 min. Primary fixation was in glutaraldehyde (1 mL, 2.5%) for 10 min. Secondary fixation was with osmium tetroxide (1%, 250 μL) for 15 min. Cells were washed twice for 15 min each after both fixation steps. Stepwise dehydration was with ethanol at 70%, 90% and 100% for 10 min each and 100% for 15 min. Samples were washed in 1 mL propylene oxide for 15 min before infiltration on a rotary mixer for 1 h with 1:1 v/v mix of propylene oxide and Procure 812 resin. This was replaced with 1:2 v/v mix of propylene oxide and resin for 16 h, then with 100% resin for 2 h before transferring to warm embedding capsules (size 00) and replacing with final 100% resin for curing at 60°C for 48 h. Room temperature blocks were trimmed and sections cut using an ultramicrotome (Ultracut, Leica). Sections were collected on 200 μm copper grids, stained with 2% uranyl acetate for 20 min, washed in CO2-free water and stained with lead citrate for 10 min. Sections were viewed at 80 kV (100SX, JEOL) at between 5000× and 40,000× magnification. Three sections per specimen were examined in at least five fields of view and photographs recorded on film.
2.6. Chlamydia trachomatis Infection Assay in McCoy Cells
McCoy cells (ATCC, CRL-1696) and a clinical isolate of C. trachomatis (RCH, Melbourne, Australia) were employed in methods based on Suchland et al. [48] and general methods for Chlamydia cell assays. Cell monolayers were infected and chlamydial bodies detected by immunofluorescent staining. NPBD was assayed to determine whether it had an inhibitory effect on infection of McCoy cells by elementary bodies (EBs) and whether it inhibited replication of reticulate bodies (RBs) in inclusion vacuoles. McCoy cell monolayers were prepared and maintained in growth medium (Minimum Essential Medium with Earle’s Salts, 7.5% w/v sodium bicarbonate, 200 mM glutamine, non-essential amino acids, 100 mM sodium pyruvate, 10% v/v foetal calf serum). Cells were grown to a confluent layer in a 175 cm2 culture flask, washed with PBS, harvested with trypsin/EDTA and resuspended in growth medium. A 96-well cell culture plate was seeded at 3 × 105 cells/mL in 200 μL per well. Cells were grown to confluency for 72 h before infection with C. trachomatis. All incubations were performed at 37°C in 5% CO2. A suitable infectious dose of C. trachomatis was determined by titration in Chlamydiamaintenance medium (growth medium as above with 0.5 µg/mL cycloheximide and 4.5g/L glucose). A concentration of Chlamydia 100× higher than the lowest dilution showing no fluorescence was used as the inoculum to provide a minimum of 80% cell infection rate. A 10-fold titration series of the C. trachomatis stock was also included in each assay plate to verify the rate of infection. The medium was aspirated from the 96-well microplate containing confluent McCoy cells and 100 µL of C. trachomatis was added to triplicate wells. The plate was centrifuged (50 min, 340 ×g, 35°C) to adsorb Chlamydia to McCoy cells. The medium was aspirated, replaced with 200 µL maintenance medium and incubated 48 h before fixing cells and staining as described below.
To test for inhibition of infection, medium was aspirated from the plate of confluent McCoy cells and 100 μL each of Chlamydia and either NPBD or tetracycline added. Plates were centrifuged (50 min, 340 ×g, 35°C). The medium (200 µL) was aspirated and replaced with 100 μL maintenance medium plus 100 µL containing 2× NPBD or tetracycline. Test concentrations were 2-fold in the ranges 0.5-5 µg/mL for NPBD and 0.06-1 µg/mL tetracycline. Plates were incubated for 48 h. All cells were fixed and stained with fluorescein-conjugated monoclonal antibodies to C. trachomatis outer membrane protein (Bio-Rad) in situ at 48 h post-infection. The medium was aspirated and wells air-dried for 2 min. Cells were fixed at -20°C for 15 min in acetone:methanol (1:2). The fixative was aspirated and cells air-dried for 3 min at room temperature. C. trachomatis antibody was added and the plate incubated for 1 h in humid conditions. Wells were washed five times in PBS/1% Tween20, once in distilled water and air-dried for 2 min. Fluorescent mounting medium (0.3M KI2, 0.3M I, 50% glycerol) was added to wells and cytopathic effect recorded semi-quantitatively. Infection of the monolayer was assessed by examining fluorescence levels under an inverted microscope (IX71, Olympus). Assays were repeated twice and digital photographs taken (A525, Canon) using an eyepiece attachment. The MIC was defined as 2-fold above the concentration resulting in alterations to 90% of inclusions [49]. NPBD and tetracycline were also tested using the method described above, except, the addition of drugs was delayed until 24 h after infection to determine whether drugs inhibited replication of RBs in inclusion vacuoles.
2.7. Yersinia enterocolitica Adherence & Invasion In Vitro Assay
Vero cells (ATCC CCL-81) were maintained in culture medium (DMEM, 10% newborn calf serum, NCS, with Penicillin/Streptomycin). Test wells were duplicated in two sets of plates per assay. Seeding density was 4×105 cell/mL in 500 µL assay medium (DMEM, 10% NCS) per well on each 24-well cell culture plate. Plates were incubated for 24 h before infection with bacteria at 37°C in 5% CO2. The test strain was Yersinia enterocolitica (ATCC 70020) and NPBD test concentrations were 2, 8 and 32 µg/mL. Salmonella enterica Typhimurium LT2-9121 (ATCC 70020) and E. coli (ATCC 25922) were used as controls for invasion levels and received no NPBD. Duplicate wells were prepared with 200 µL of 2×NPBD in assay medium with 2% DMSO. Overnight bacterial cultures in MHB were diluted 1/100 in assay medium and incubated for 2h, centrifuged at 5000×g for 2 min, resuspended in assay medium to ~1×108 CFU/mL and incubated 15 min. Wells were overlaid with 200 µL bacteria, mixed gently and returned to incubate. After 1.5 h incubation, one plate was used to estimate the number of adherent and intracellular bacterial cells. Cells were washed three times with PBS and lysed with 400 µL of 0.1% Triton-X100 for 15 min. PBS (600 µL) was added and the total bacteria estimated by viable counts. After 2.5 h incubation, the second plate was used to estimate the number of intracellular bacteria. Cells were washed with PBS, gentamicin (200 µL, 200 µg/mL) added and the plate incubated 1 h to kill extracellular bacteria. Wells were again washed with before lysis with Triton-X, dilution with 600 µL PBS and viable counts performed. Results below are an average of three independent assays. The number of adherent bacterial cells was calculated by subtracting the number of intracellular cells from the total number of intra- plus extracellular cells.
2.8. Prodigiosin Synthesis Assay in Serratia marcescens
Prodigiosin production in S. marcescens (RMIT 342/1-32) was evaluated by modification of the method used by Dierstein et al. [50]. S. marcescens was grown for 6 h in 30 mL peptone glycerol broth (PGB), cells concentrated by centrifugation (10 min, 5000 ×g; 3K15, Sigma), resuspended in 6 mL sterile distilled water, and stored overnight at room temperature. The suspension inoculum was diluted in PGB to A660nm of 0.05 and 15 mL divided into each of four 50 mL culture bottles. NPBD (15 µL) was added to 3 bottles at 8, 32 and 128 µg/mL. All, including the control contained 1% v/v DMSO. Cultures were incubated at 30°C, optimal for pigment production. At NPBD addition (0 h), and 4 h, 8 h and 12 h, 300 µL was removed into 2.7 mL methanol with 4% 1 N HCl at room temperature for 1 h to extract the pigment. The solution was clarified by centrifugation at 5000 ×g for 15 min and A500nm used to measure the extracted pigment. Percentage inhibition of pigment production relative to the control in the absence of NPBD was calculated OD600nm measurements or viable counts were performed at time points to verify levels of growth over three separate assays.
2.9. Swarming Motility Assay in Proteus spp.
Luria broth agar (LBA) plates were prepared based on the method of Liaw et al. [51] containing 1% agar to facilitate swarming of Proteus mirabilis and P. vulgaris. NPBD in DMSO was added immediately before pouring to give 2-fold concentrations of NPBD between 4 and 64 µg/mL and 1% v/v DMSO. Control plates received 1% v/v DMSO alone. Plates were prepared fresh and dried at 37°C for 30 min prior to inoculation. A colony from overnight LBA plates of each species was used to inoculate MHB and incubated for 6 h. LBA plates containing NPBD at 0, 4, 8, 16, 32 and 64 µg/mL were centrally inoculated with 5 µL of culture and incubated aerobically for 16 h. To explore the effect of NPBD exposure on swarming motility prior to surface growth, LBA plates were prepared as above without NPBD. An overnight plate of each species was used to inoculate 5 mL MHB containing NPBD at 8, 32 and 128 µg/mL and incubated for 6 or 24 h. This was used to centrally inoculate LBA plates with 5 μL and plates were incubated for 16 h.
3. RESULTS & DISCUSSION
The diverse studies presented have the objectives of elucidating whether the activity of NPBD can be attributed to any well-known bacterial targets or can be attributed to inhibition of microbial PTPs. The assays were selected to show interference with known PTP functions in bacterial species, including those which are virulence attributes of clinically significant pathogens and to investigate whether NPBD has any effect on known bacterial drug targets. The data serve to define attributes of NPBD significant to its clinical use as a potential anti-infective drug.
3.1. Inhibition of Protein Tyrosine Phosphatase Activity
NPBD was a less potent inhibitor of tyrosine phosphatases (PTP1 B and Yop) than vanadate (Fig. 2). Vanadate showed 73% and 87% inhibition at 60 nM for PTP1 B and CD45 respectively. Vanadate was much less effective against the bacterial enzyme Yop with 27% inhibition at 60 nM. At 40 µM NPBD showed 46% inhibition of PTP1 B and 42% inhibition of Yop. It did not show specific inhibition of the DSP, CD45 at up to 40 µM. There was no inhibition of protease activity by sodium orthovanadate up to 16 µM and NPBD to 40 µM.
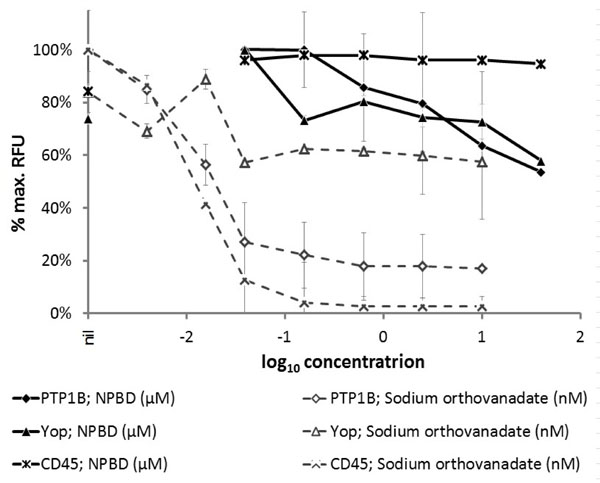
Phosphatase activity of PTP enzymes (PTP1B, 12 mU; Yop, 0.4 U; CD45, 3U) in the presence of test compound NPBD or sodium orthovanadate as a positive inhibitor control. Relative fluorescence units (RFU) are expressed as a percentage of the maximum achieved in each assay for individual enzymes ±SEM.
Sodium orthovanadate is a broad and highly active phosphatase inhibitor, as would be expected of a phosphate analog, and widely used as a PTP positive inhibitor control in enzyme assays. It has been investigated as a possible anti-tumor agent [52, 53]. Benzyl-β-nitroethene and derivatives have also been shown to be inhibitors of protein tyrosine phosphatases PTP1B and Yop [10, 12]. NPBD, a tyrosine mimetic, showed selective inhibition of PTP1B and Yop but not the serine/threonine phosphatase, CD45, at the highest concentration tested. PTP1B is a classical, non-transmembrane PTP involved in many cellular processes such as apoptosis, cell-to-cell adhesion, motility and invasion, including regulation of cortactin tyrosine phosphorylation [54]. It has been shown to be a regulator of signalling involved in human diseases and has become a target in the search for drugs to treat cancer, obesity and type 2 diabetes [55-57]. PTP1B has been shown to both promote and suppress tumorigenesis [56]. Benzyl nitroalkenes have been reported as having anti-tumor activity [58-60]. Leite et al. [61] reported that 1,3 benzodioxole petidyl derivatives inhibited sarcoma in albino mice. NPBD (2 µM) selectively inhibited a human non-small-cell lung cancer cell line (A549) in the SRB assay [62]. NPBD did not inhibit CD45 at 40 µM. CD45 is a receptor-linked PTP found on all non-nucleated haematopoietic cells and critical to T-cell and, to a lesser extent, B-cell function. CD45 has a role in immuno-receptor modulated signalling by regulating src PTKs and inhibition of CD45 would have implications for host immunity [63].
3.2. Bacterial ATP Synthesis
ATP production correlated with increased cell numbers for untreated B. cereus and S. aureus (Fig. 3). Exposure to NPBD showed no greater effect on ATP production than could be accounted for by a corresponding drop in the number of metabolizing cells. Cells in this assay are growing exponentially and nearly all cells would be viable and producing ATP by oxidative phosphorylation, so measurements would be predominantly intracellular ATP generated by membrane-bound ATP synthase. NPBD does not disrupt cellular membranes and there is no evidence from this assay that NPBD is directly affecting ATP synthase involved in aerobic ATP production. This is consistent with the observation that NPBD is equally inhibitory to facultative aerobic energy producers when cells are forced into fermentative energy production under anaerobic conditions (Nicoletti et al., unpublished). F0F1ATP synthases are the terminal enzymes of the oxidative phosphorylation pathway responsible for the majority of ATP synthesis [64] and are highly conserved across mitochondria and bacterial plasma membranes [65]. Several tyrosine phosphoproteins have been identified in the sub-units of ATP synthase [66, 67]. A tyrosine mimetic might be expected to inhibit this enzyme but NPBD does not appear to significantly affect bacterial ATP synthesis in B. subtilis or S. aureus. In a microarray preliminary analysis of the effect of NPBD on gene expression in B. subtilis, expression of genes encoding ATP synthase subunits were down-regulated, only at the highest concentration (16 µg/mL) [12]. A variety of compounds, including peptides and antibiotics inhibiting ATP synthase are reviewed by Hong and Pedersen [68].

Effect on total ATP production (nM) by B. cereus (a) and S. aureus (b) in MHB exposed to NPBD (µg/mL). ATP (―) and viable cell numbers (- - -) were measured at each time point by the BacTitre-Glo™ ATP assay and viable counts respectively. B. cereus exposed to 0 (×), 16 µg/mL (■) and 32 µg/mL (▲) NPBD and S. aureus exposed to 0 (×), 8 µg/mL (■) and 32 µg/mL(▲) NPBD. Regression analysis of ATP production and CFU/mL for increasing treatment concentrations gave R2 values of 0.94, 0.75 and 0.91 for B. subtilis respectively; and R2 values of 0.79, 0.74 and 0.67 for C. albicans respectively.
3.3. RNA Transcription and Translation
The E. coli S30 extract system allows in vitro investigation of ribosomal activity which can be used for screening compounds for inhibition of protein synthesis. There was no change in the rate of transcription or translation of β-galactosidase (measured by hydrolysis of ONPG to ONP) at NPBD up to 16× the MIC (Fig. 4). Slopes indicating the rates of protein synthesis for all test reactions averaged 0.98 with a standard deviation of 0.01. There was no interference by NPBD with absorbance readings of ONP in the absence of pGEM-βgal DNA. Absorbance readings during the assay were elevated only due to the absorbance of NPBD at the highest concentrations of 64 and 128 µg/mL. No concentration-dependent effect of NPBD on the rate of the reaction was observed, indicating no effect on cell-free protein synthesis. This supports the observation of a lack of interaction (synergistic or antagonistic) between NPBD and erythromycin or tetracycline [12] which affects different stages of protein synthesis. In bacteria, transcription is accomplished by RNA polymerase (RNAP) with a conserved core linked to a sigma subunit that specifies binding to promoter elements and transcript regulators. In E. coli, many additional factors modify RNAP activity [69] and bacterial RNAPs show considerable variation in their domains across prokaryotic lineages [70, 71]. Extracted enzyme systems from one species, therefore, may not be as sensitive in detecting inhibition of the RNAP of another species. Reports using assays based on extracted enzyme systems suggest variable responses to inhibitors by E. coli, S. aureus and Streptococcus pneumoniae [14, 15]. While indicative, the failure of NPBD to inhibit transcription or translation in E. coli should be prudently extended to other bacterial species.
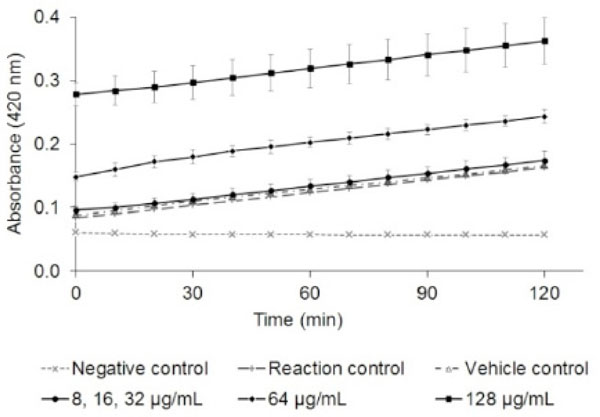
Effect of NPBD (µg/mL) on cell-free synthesis of β-galactosidase by measurement of ONP (A420nm) ±SD. The relative rate of protein synthesis (slope of the curve with NPBD / slope of the reaction curve at the maximum rate of protein synthesis without NPBD) was the same for all concentrations of NPBD (average 0.98, ±0.01).
3.4. Integrity of the Cell Wall and Cytoplasmic Membrane
The effect of NPBD on the integrity of the cell wall and cytoplasmic membrane was assessed respectively by lysis of intact cells of C. albicans and B. cereus and by lysis of spheroplasts of E. coli and M. catarrhalis.
In the whole cell lysis assay a reduction in viable cells without loss of turbidity indicates non-lytic microbicidal activity. Exposure of B. cereus and C. albicans cultures to NPBD (64-128 µg/mL) shows a similar concentration-dependent rate of kill over 8 h with a 3-log reduction in viable cells for both species without a corresponding decrease in turbidity (Fig. 5). Non-lytic action against B. cereus and C. albicans is confirmed in the ultrastructure study below and is likely to apply generally to Gram positive bacteria and yeast.
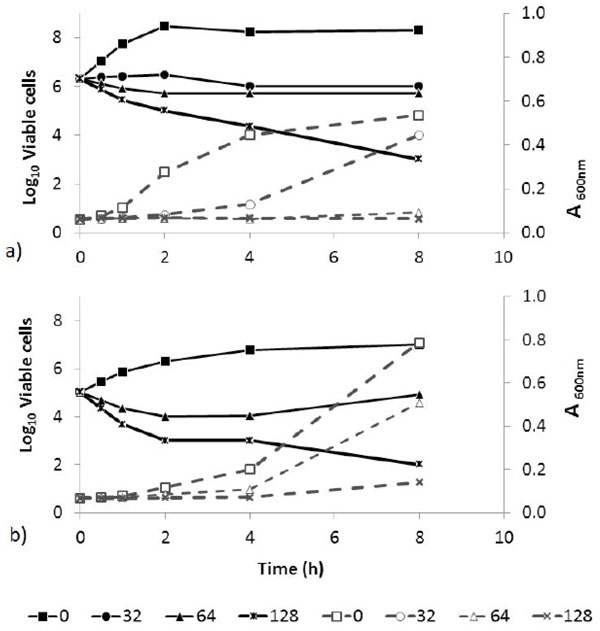
Absorbance (600nm; - - -) and viable counts (log10 CFU/mL; ―) of B. cereus (a) and C. albicans (b) over 8 h exposure to NPBD (µg/mL). Microbicidal activity with no proportional loss of turbidity is seen with the highest NPBD concentrations above the MIC (8 µg/mL) for both species.
Cell lysis can be due to disruption of the cytoplasmic membrane or secondary to disruption of the cell wall. Spheroplasts, lacking the protection of the cell wall or outer membrane (OM), are highly sensitive and their disruption or degradation indicates a direct action on the bacterial cytoplasmic membrane. NPBD is more rapidly microbicidal against E. coli than M. Catarrhalis [12]. Over 60 minutes there was no decrease in the absorbance of M. catarrhalis spheroplasts or intact cells with NPBD at concentrations equal to or 2-fold above the MIC (16 μg/mL) indicating no degradation of either cell (data not shown). For E. coli, a dose-dependent decrease in absorbance was initially more rapid and remained stable to 60 min for both spheroplasts and intact cells (Fig. 6). The lack of increased susceptibility of E. coli spheroplasts compared to intact cells indicates that NPBD does not lyse spheroplasts primarily or secondary to degradation of the outer membrane. The similarity between bacterial and yeast plasma membranes indicates NPBD is unlikely to disrupt membranes generally. NPBD appears not to degrade Gram cell envelope or cell walls of B. cereus and C. albicans.

Percentage of Escherichia coli spheroplasts and intact cells on exposure to NPBD (µg/mL). Turbidity was monitored by OD570nm every 10 min for 1 h. The percentage of intact spheroplasts or whole cells was calculated as 100 x (A570 nm at time X / A570 nm at time 0).
3.4.1. Permeation of the Gram-Negative Cell Envelope
NPN fluoresces weakly in aqueous environments and strongly in a hydrophobic phospholipid bilayer [72]. The outer membrane (OM) of Gram-negative bacteria is a significant permeability barrier protecting the cytoplasmic membrane. When the OM is disrupted by an antimicrobial agent, the fluorescent probe NPN can partition into the hydrophobic core of the OM. NPN uptake by E. coli exposed to NPBD, EDTA and polymyxin B is shown in (Table 1). EDTA and polymyxin B, known OM disrupters, resulted in NPN-uptake values of 1.4× and 2.9× that of the negative control. NPBD, at 250 µg/mL for 5 min, failed to increase fluorescence or NPN-uptake values above that of the negative control, indicating no change in permeability of the OM. NPBD is a small, highly lipophilic, neutral molecule which can permeate the outer membrane of E. coli to inhibit growth but does not damage the cell wall or membrane.
Change in permeability of E. coli exposed to NPBD for 5 min as measured by uptake of fluorescent NPN into the outer membrane lipid bilayers.
^ Average RFU were calculated by subtracting fluorescence of identical wells without NPN,
* NPN-uptake value is a ratio of relative fluorescence of the test well containing NPBD, Polymyxin B or EDTA, and the relative fluorescence of the control well containing HEPES buffer alone. EDTA and polymyxin B are positive controls which disrupt the OM allowing penetration of NPN into the lipid bilayer.
3.4.2. Activity Against Lipopolysaccharide Deficient Campylobacter jejuni
NPBD has low activity against many enteric Gram-negative bacteria possessing high density LPS or lipo-oligosaccharide (LOS) outer membranes which form an effective barrier to lipophilic drugs [73]. Mutants lacking LPS or LOS have been found to lose resistance to lipophilic antibiotics [74, 75]. NPBD has relatively low activity against Campylobacter spp. with MIC in the range 32-256 µg/mL (Nicoletti et al., unpublished). NPBD is a highly lipophilic molecule and its relative inactivity against many Gram-negative species could be partly due to its exclusion by the outer membrane. The mean MIC for NPBD against C. jejuni wild type (HB9313) and a LOS-deficient mutant (WaaF–) were each 256 µg/mL. The severely truncated LOS structure in the WaaF– mutant did not result in a greater susceptibility to NPBD indicating that the presence of intact LOS in the outer leaflet of the OM was not a barrier to entry of NPBD, confirming its ability to permeate the Gram-negative OM shown in the NPN assay.
3.5. Effect of NPBD on Ultramorphology
No significant morphological changes were observed in B. cereus or C. albicans cells exposed to NPBD, examined at up to 40,000× Figs. (7, 8). There was no evidence of swollen cells, thickened, incomplete or defective walls, abnormal septation or detached or undulating cytoplasmic membranes. The resolution was not adequate for detection of changes in sub-cellular structures, however, cytoplasmic aggregates or uneven staining were not observed. The lack of effect on the structural integrity of the cell walls and cytoplasmic membranes of B. cereus and C. albicans confirms the lack of lytic effect on cell walls and cell membranes shown in other assays above. A noted effect was the much lower incidence of endospores in NPBD-treated B. cereus compared to control samples. C. albicans treated cells appeared morphologically similar to control cells and showed normal budding (Fig. 8). The inhibition of sporulation observed here is supported by a whole genome transcription expression analysis to determine the concentration-dependent effects of NPBD (2 and 16 µg/mL for 30 minute exposure) on B. subtilis [12]. The most down-regulated gene, spoVG, was decreased 7-fold and 17-fold in 2 and 16 µg/mL treatments respectively. The spoVG gene is associated with cell division and initiation of sporulation [76]. Phosphorylation of spoOA on aspartate regulates activity [77] however, to date no involvement of tyrosine kinases or phosphatase in endospore formation in Bacillus spp. has been reported. NPBD could be indirectly affecting sporulation by interfering with one or more signals for the initiation of cell septation and endospore formation.
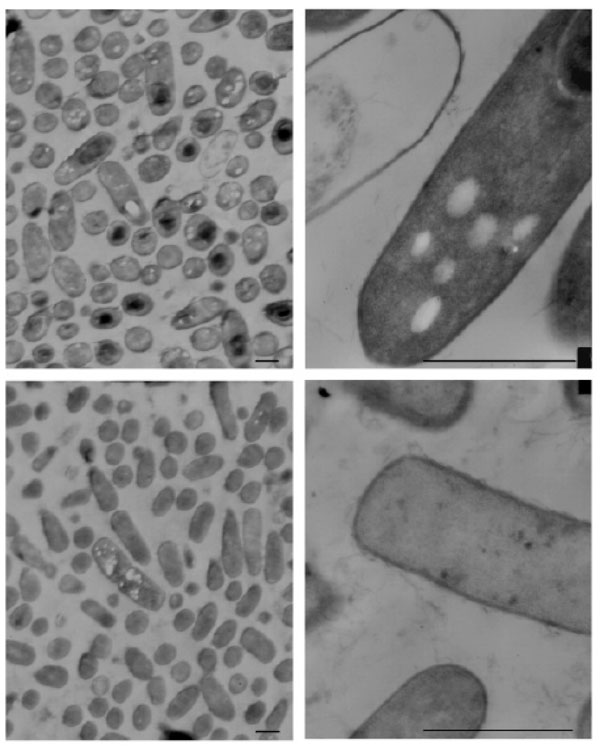
Transmission electron micrograph of Bacillus cereus. Top panel: B. cereus untreated. Bottom panel: B. cereus exposed to 2 μg/mL NPBD for 24 h. Scale bars represent 1.5 µm.
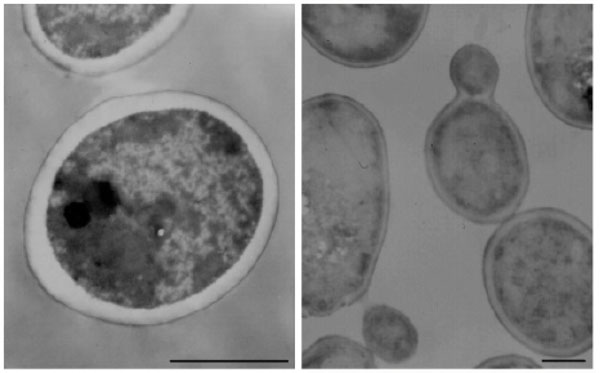
Transmission electron micrograph of Candida albicans. Left: C. albicans untreated. Right: C. albicans exposed to 4 μg/mL NPBD for 24 h. Scale bars represent 1.5 µm.
3.6. Inhibition of Chlamydia trachomatis Infection in McCoy Cells
Chlamydia have a biphasic development cycle, existing as two functional and morphological forms over the life cycle of 48-72 h. Following host cell entry, the chlamydial developmental cycle occurs entirely within an intracellular inclusion body. Elementary bodies (EBs) are small, metabolically inactive, extracellular infectious forms which bind to host cells inducing endocytosis, the formation of intracellular inclusions and cytoskeletal reorganization [78]. The inclusion membrane contains the chlamydial outer membrane complex which is responsible for EB integrity and interaction with host cell membranes [79, 80]. Within the inclusion body, EBs differentiate into larger metabolically active, non-infectious reticulate bodies (RBs) within 6-8 h. RBs replicate by binary fission using energy and biosynthetic precursors from the host cell and redifferentiate into EBs which are released by cell lysis or extrusion of the inclusion to continue the infection cycle [49].
NPBD and tetracycline were administered at the time of infection of McCoy cells and 24 h after infection to investigate any differential effect on the entry of EB and formation of inclusion bodies and the later replication of RB in inclusion bodies. NPBD inhibited C. trachomatis infection when added at the time of inoculation of the McCoy cell monolayer with EBs. NPBD (2.5 and 5 µg/mL) showed a concentration-dependent reduction in the number of reticulate bodies at 48 h comparable to the reduction by tetracycline (Fig. 9). Applying the method of Stephens et al. [49], the MIC for NPBD against Chlamydia is 10 µg/mL. Results for tetracycline (MIC 0.25 μg/mL) were comparable to those reported in Suchland et al. [48]. NPBD administered after the formation of RB, 24 h post-infection, did not inhibit their replication while tetracycline did produce a reduction at a higher treatment concentration. NPBD could be interfering with binding and/or entry within the first few hours of infection but is not able to inhibit replication in inclusion bodies at the maximum concentration used in this assay. When administered in the later stages of chlamydial infection NPBD had no inhibitory effect on RBs suggesting no interference with chlamydial or commandeered host cell metabolic functions involved in chlamydial replication.
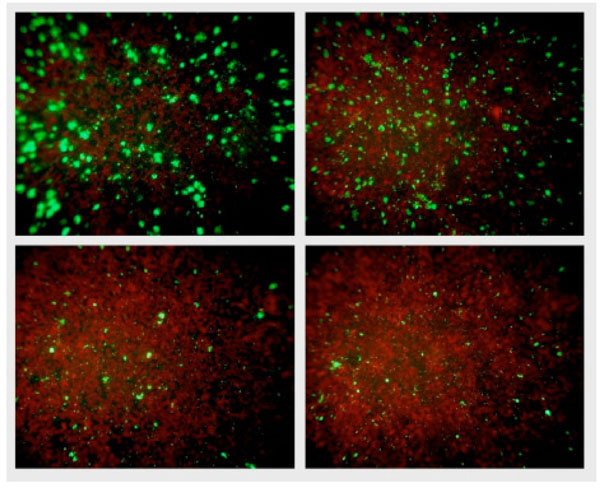
Fluorescent chlamydial bodies 48 h after infection. Clockwise from top right: Untreated McCoy cells, and McCoy cells exposed to 2.5 μg/mL NPBD, 5 μg/mL NPBD and 0.06 μg/mL tetracycline administered at the time of infection.
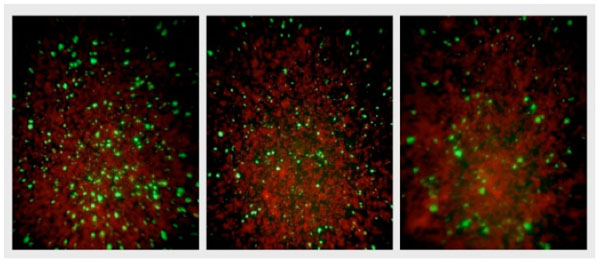
Fluorescent chlamydial bodies 48 h after infection of untreated McCoy cells (Left), and McCoy cells exposed to 5 μg/mL NPBD (middle) and 0.5 μg/mL tetracycline (right) administered 24 h after infection.
There are several metabolic targets in chlamydial infection that a PTP inhibitor might affect. C. trachomatis has two protein kinases, CTPkn5 and CTPknD, but no open reading frames for likely PTPs have been identified [81]. Therefore, any inhibition is likely to be of host cell PTPs. Chlamydia exploit host cell signalling and trafficking pathways for invasion and development. Chlamydial infection induces tyrosine phosphorylation of chlamydial and host proteins very early in infection consistent with a role in pathogen entry [41, 42]. Entry of C. trachomatis EBs is accompanied by tyrosine-dependent phosphorylation of host cytoskeleton-related proteins [79, 82]. The focal adhesion kinase (FAK) and the docking protein SHC are tyrosine phosphorylated at the time of C. pneumoniae attachment and entry [29]. Chlamydia possess a type III secretion system (TTSS), as do many intracellular pathogens. The TTSS is a protein export apparatus allowing microbial proteins to be secreted across the cytoplasmic membrane into the cytosol of eukaryotic cells to modify host cell processes involved in cell invasion, to modify the inclusion membrane and host regulatory pathways [83]. A chlamydial trans-located actin-recruiting protein (TARP) is a tyrosine phosphoprotein translocated into the host cell by the chlamydial TTSS [41, 84]. TARP is rapidly phosphorylated by host kinases and is important in actin recruitment at the entry site, formation of the EB inclusion and cytoskeletal reorganization [84]. The tyrosine phosphorylated domain of TARP, however was shown not to be associated with actin recruitment [85]. Swanson et al. [86] report species-specific induction of ezrin tyrosine phosphorylation in C. trachomatis. Ezrin, a member of the ezrin-radixin-moesin (ERM) protein family, serves as a link between host cell receptors and the actin cytoskeleton and is essential in pathogen entry. Chlamydial infection activates host tyrosine kinases, such as the Src family which have been reported to play a key role in C. trachomatis intracellular microtubule trafficking of the inclusion vacuole and in the chlamydial development cycle [87, 88]. There are thus several possible targets for NPBD to explain the inhibition of entry of C. trachomatis into mammalian cells. In Chlamydia, PTP profiles vary between species and within infecting strains [41, 42]. Therefore, extrapolation of data between Chlamydia species is problematic. Inhibition of chlamydial entry and subsequent formation of RBs supports the contention that NPBD could be an effective agent against intracellular pathogens. NPBD (10 and 25 µg/mL) has been shown to be effective in inhibiting growth and replication of Plasmodium falciparum 3D7 in human red blood cells [11].
3.7. Inhibition of Yersinia enterocolitica Adherence & Invasion In Vitro
NPBD inhibited Yop in an enzyme assay and this inhibitory effect is confirmed in a cell model of invasion of Vero cells by Y. enterocolitica. NPBD is much more active against Y. enterocolitica (MIC 16 µg/mL) than against most other enteric Gram-negative bacilli. Treatment with 8 µg/mL NPBD reduced the number of adherent cells of Y. enterocolitica by 40% and at 32 µg/mL by 66% (Fig. 11). Only 1% of adherent Y. enterocolitica cells invaded Vero cells and only 0.01% invaded in the presence of 32 µg/mL NPBD. NPBD at 2, 8 and 32 µg/mL reduced invasion of adherent cells by 44%, 98% and 99% respectively. Cell invasion by Salmonella LT2, was 3.5%, and by E. coli was 0.2%.

Average CFU/mL of Y. enterocolitica, E. coli and S. Typhimurium LT2, for Adherence (Adh) or Invasion (Inv) in Vero cell assays when exposed to NPBD at 2, 8 and 32 μg/mL (±SEM).
Yersinia possess YopH, a PTP belonging to a family that targets tyrosine phosphoproteins in eukaryotic cells. YopH targets focal adhesion proteins involved in invasin-mediated uptake and facilitates entry by disrupting cytoskeletal structure and host cell regulation [22, 89]. Inhibition of YopH would be expected to reduce the number of adherent and intracellular bacterial cells. The Yersinia adhesins YadA and invasin mediate attachment, entry and colonization of mammalian epithelial cells by binding to β1 integrins, serine/threonine phosphoprotein transmembrane receptors [90, 91]. YadA or invasin mediated invasion of hepatic cells by Y. pseudotuberculosis was impaired by inhibitors of tyrosine, serine/threonine, and phosphatidylinositol 3 (PI3) kinases [90]. In Y. pseudotuberculosis non-receptor protein tyrosine kinases play a significant role in YadA-mediated cell processes [92]. Tyrosine phosphorylation of Phospholipase C-γ1, a downstream effector of the serine/threonine signaling protein PI3, is required for efficient cell invasion by Y. pseudotuberculosis [93]. NPBD may be interfering with the signalling of one or more tyrosine kinases or phosphatases in addition to inhibition of YopH. Several inhibitors of YopH have been reported [39, 94, 95].
3.8. Inhibition of Prodigiosin Secretion by Serratia marcescens
NPBD had a marked effect on prodigiosin production by S. marcescens both observed visually and by absorbance (A500nm) of pigment extracts (Fig. 12). Maximum inhibition of pigment productionin S. marcescens exposed to NPBD occurred at 8 h with 165% less extracted pigment compared to the untreated control. This inhibition occurred at a concentration of 128 µg/mL (4-fold below the MIC) suggesting inhibition may be quite specific. There was a dose-dependent response between 8 µg/mL and 32 µg/mL (153% and 164% inhibition at 8 h respectively). Cell proliferation and inhibition of pigment production peaked at 8 h and reduced inhibition of pigment production for 8, 32 and 128 µg/mL was observed at 12 h (59%, 91% and 98% inhibition respectively). This is consistent with production patterns of secondary metabolites during the stationary phase and an increase in production can explain the reduction in inhibition, though the level of inhibition remains high. At maximum cell density, the multiple cellular and environmental cues which are involved in the formation or secretion of prodigiosin may outweigh the inhibition of one or more PTPs by NPBD.

Inhibition of prodigiosin produced by Serratia marcescens exposed to NPBD. Panels (from top to bottom) show cultures at time points 0, 4, 8 and 12 h respectively. Cultures in each panel contain treatment concentrations of 0, 8, 32 and 128 µg/mL NPBD respectively (left to right).
Prodigiosin is produced by some Serratia species and actinomycetes and is a secondary metabolite, appearing only in the later stages of bacterial growth [96, 97]. Whether prodigiosin is a significant virulence factor in Serratia is not yet clear, since non-pigmented strains are isolated from clinical specimens [98]. Prodigiosin has antibacterial, anti-fungal, anti-coccidial and anti-malarial activity, a competitive advantage that may contribute to virulence [99, 100]. Fürstner et al. [101] have shown that nonylprodigiosin is a PTP and DSP inhibitor, with greater activity against PTP. The prodigiosin biosynthetic pathway has been elucidated and requires gene products from the pigA-pigO operon [102-104]. Very few tyrosine phosphoproteins have been identified in the production of the prodiginenes. Li and Strohl [105] isolated ptpA, encoding allow molecular weight PTP, from Streptomyces coelicolor. PtpA belongs to a highly conserved family that have closely related homologues in prokaryotes [105, 106]. Overexpression of ptpA in Streptomyces lividans increased the production of undecylprodigiosin [107]. The high level of inhibition of early stages of prodigiosin production by NPBD would suggest that some as yet unidentified tyrosine phosphatases have been targeted.
3.9. Inhibition of Swarming Motility in Proteus spp.
Motility, in its many forms, is a significant virulence factor in many bacterial species. Swarming is a flagella-dependent surface translocation involving rapid movement of ‘swarmer’ cells across a semi-solid surface, facilitating colonisation and biofilm formation [108]. It is a coordinated social behaviour involving cellular communication and regulation by quorum sensing and flagellar regulator genes [108]. For Proteus spp., a characteristic periodic pattern of concentric circles is produced on plate cultures from the point of inoculation due to the repetitive alternation between growth of stationary cells (consolidation) and the production and subsequent migration of specialised swarmer cells possessing up to 50 times as many flagella [109]. Swarming is facilitated by the secretion of peptide surfactants such as in P. mirabilis which secretes a capsular polypeptide that reduces surface friction [110]. Exposure during surface growth on LBA incorporating NPBD investigated the effect of NPBD on the characteristic swarming patterns of Proteus spp.
NPBD caused a dose dependent inhibition of swarming in both P. mirabilis and P. vulgaris (Fig. 13). P. mirabilis with 64 µg/mL NPBD and P. vulgaris with 32 µg/mL NPBD grew as confluent buttons with total suppression of swarming. Treatment with NPBD (8, 32 and 128 µg/mL) for 6 h or 24 h prior to inoculation on LBA resulted in concentration-dependent inhibition of swarming for P. vulgaris, with swarming almost completely inhibited by pre-exposure to 32 µg/mL for 6 h (Fig. 13). Pre-exposure to NPBD for 6 h did not affect cell viability or swarming of P. mirabilis and exposure for 24 h only inhibited swarming at 128 mg/mL (Fig. 14).
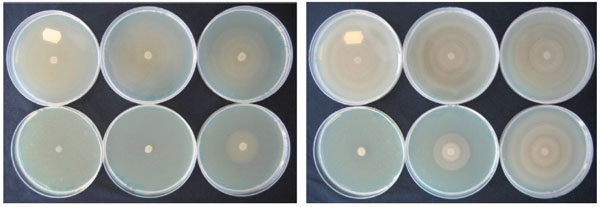
Effect of NPBD on swarming motility of P. vulgaris (left) and P. mirabilis (right) in LBA containing 0, 4, 8, 16, 32, 64 µg/mL NPBD (clockwise from top left ) centrally inoculated with log phase culture and incubated aerobically at 37°C for 16 h.
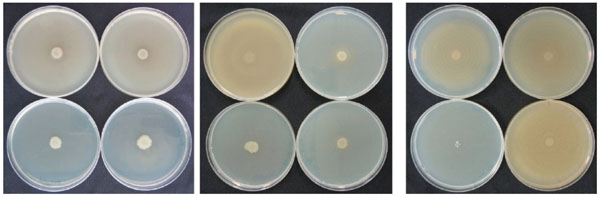
Effect of NPBD on swarming motility of P. vulgaris exposed in broth culture for 6h (left) or 24h (middle) and P. mirabilis for 24h (right) prior to plating on LBA. NPBD concentrations clockwise from top left in each photograph: 0, 8, 32, 128 μg/mL.
The standard MIC of NPBD for P. mirabilis is 128 µg/mL and for P vulgaris is 16 µg/mL [5]. NPBD inhibited swarming in P. mirabilis at 64 µg/mL and in P. vulgaris, at 32 µg/mL when NPBD was continuously present in the medium during surface growth. The 8-fold difference in toxicity between P. mirabilis and P. vulgaris does not reflect the difference in concentrations required to damage the pathways involved in swarming motility. The metabolic target of NPBD in these pathways may be more susceptible or more significant in P. vulgaris.
Swimming motility is the rapid movement of individual bacterial cells through an aqueous or semi-soft solid medium (<0.3% agar) driven by rotation of one or more flagella and is regulated by the flagellar master. NPBD did not affect swimming motility in Proteus as assessed by a semi-solid liquid assay (data not shown). Therefore, NPBD may be interfering with signaling pathways involved in quorum sensing. Tyrosine phosphorylation has been found to be involved in similar signaling events [19, 111], though a specific role for tyrosine phosphoproteins in motility has not been identified. NPBD inhibition of swarming motility suggests that an unidentified PTP could be involved in Proteus spp. Ferreira et al. [112] showed that BceF proteins from Burkholderia cepacia belonging to the bacterial tyrosine (BY) kinase family are involved in stress responses and virulence. A comparative transcriptome profile between a bceF mutant and the wild-type B. cepacia showed that genes with decreased expression in the bceF mutant were related to reduced swimming and swarming motility and alteration in biofilm architecture [113].
CONCLUDING REMARKS
Inhibition of PTPs is most likely the major mechanism of action of NPBD in microorganisms. The compound does not interfere with major metabolic functions in bacteria that are the targets of major classes of anti-infective agents, namely, cell wall and cell membrane integrity, RNA translation and transcription and ATP production. Inhibition of the early stages of the development cycle of C. trachomatis confirms NPBD acts intracellularly, probably by interference with host PTPs. NPBD inhibits several bacterial virulence factors in which PTPs have been shown to be involved; attachment and invasion by Yersinia spp., motility in Proteus spp., pigment production in Serratia spp. and endospore formation in Bacillus spp. An agent that kills significant human pathogens, inhibits intracellular pathogen replication and additionally inhibits virulence mechanisms aiding entry and infection would be a desirable anti-infective drug. These studies suggest that NPBD is a good candidate for drug development, particularly for infections that initiate or occur at mucosal surfaces. NPBD is a lipophilic, neutral tyrosine mimetic which rapidly permeates cell membranes, in contrast to many known PTP inhibitors which are hydrophilic and penetrate cell poorly. NPBD is active at µM concentrations, can be effective against intracellular pathogens and has an acceptable toxicity profile. Some toxicology data has been reported [5, 11, 13].
PTPs provide a much needed new drug target in microorganisms. The catalytic site of PTPs is highly conserved which makes it difficult to achieve selective inhibition of any target PTP. Selectivity of action as an anti-bacterial drug is more likely given the structural and functional differences between prokaryotic and eukaryotic PTPs. While PTK inhibitors are successful cancer drugs there are as yet no PTP inhibitor drugs. One PTP may regulate several pathways or one pathway may be co-regulated by several PTPs. PTP inhibitors may thus have the potential for unwanted side effects. A probable advantage for a PTP inhibitor is that the development of microbial -resistance in vivo is likely to be slow since it acts on multiple targets and multiple mutations would be required for significant resistance to develop. Our results suggest that NPBD is inhibiting several PTPs to differing degrees and that the PTP targets can vary between species indicating its potential usefulness as a pathogen-specific drug in addition to a broadly active drug. The beneficial and adverse effects of inhibitors on cellular activity involving PTP signalling must be further investigated in both pathogen and host. The data presented here provides strong evidence that full characterisation of benzyl nitroalkenes as antimicrobial agents with PTP inhibition as a mechanism of action is warranted.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflicts of interest.
PATIENT’S CONSENT
Declared none.
ACKNOWLEDGEMENTS
The authors wish to thank R. Alexander (Royal Children’s Hospital, Melbourne, Australia) for providing C. trachomatis specimens and guidance and Dr Viraj Perera (RMIT University, Melbourne, Australia) for providing C. jejuni HB93-13 wild type and LOS-deficient mutant (WaaF-). Biodiem Ltd provided PhD scholarship support for studies reported in this article.

