All published articles of this journal are available on ScienceDirect.

C4-Substituted Isoquinolines: Synthesis and Cytotoxic Action
Abstract
A facile synthesis of the C4-substituted isoquinolines 5a-c and 6a-c is described. Commercially available 4-bromoisoquinoline is converted to the α,β-unsaturated esters 8 and 10 on treatment with the appropriate acrylate ester under Heck reaction conditions. The saturated amides 5a-c were obtained from the reaction of ester 9 with the requisite primary amine. Similarly the unsaturated analogues 6a-c were prepared by reacting ester 10 with the appropriate amine. The cytotoxicity of the target molecules was evaluated in two tumour cell lines in vitro. Two compounds, 6b and 6c, showed sufficient activity in the human non-small cell lung cancer line NSCLC-N16-L16 to be worthy of further study.
INTRODUCTION
Suitably substituted quinolines and isoquinolines constitute the principal subunits of a large number of linear and angular aromatic “frameworks” with cytotoxic properties (Langer Clin Cancer Res 1999) [1] (von Nussbaum JMC 1999) [2]. These molecules interact either with topoisomerase II or bind in the major or minor groove of DNA. In some cases they form DNA-intercalated molecular complexes, which are stabilized via hydrophobic interactions, hydrogen bonding and/or van der Waal’s forces (Morrell JMC 2006) [3]. The presence of nitrogen bearing side chain(s) in specific positions of their skeletons improves binding affinity and enhances solubility under physiological conditions (Werbel JMC 1986) [4]. Previously, we have published the synthesis of the cytotoxic pyrroloquinoline derivatives 1-3 (Fig. 1) (Vlachou Heterocycles 2002) [5] and their isomeric pyrroloisoquinolines 4a-e (Fig. 1) (Vlachou EJPS 2002) [6].
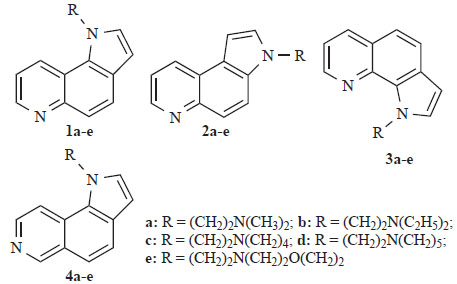
Structures of derivatives of 1H-pyrrolo[2,3-f]quinoline 1a-e, 3H-pyrrolo[2,3-f]quinoline 2a-e, 1H-pyrrolo[3,2-h]quinoline 3a-e and 1H-pyrrolo[2,3-f]isoquinoline 4a-e.
In these molecules the spatial orientation of the side chain with respect to the planar aromatic chromophore appears to influence their cytotoxic potency and DNA-binding affinity. Based on these findings we present herein the synthesis and biological activity of the C4-substituted isoquinolines 5a-c (Scheme 1) and 6a-c (Scheme 2), which constitute structurally related congeners of 1-4. For the synthetic plan we had to start with commercially available 4-bromoisoquinoline (7), which was reacted with ethyl acrylate under Heck reaction conditions (Sakamoto Heterocycles 1981) [7] to give α,β-unsaturated ethyl ester 8 in 79% yield as a single isomer of trans geometry. Catalytic hydrogenation of 8 over Pd/C in THF under 50 psi pressure led to the formation of the saturated ethyl ester 9, which was converted to the desired amides 5a-c, upon heating (110oC) with the appropriate primary amine [8].
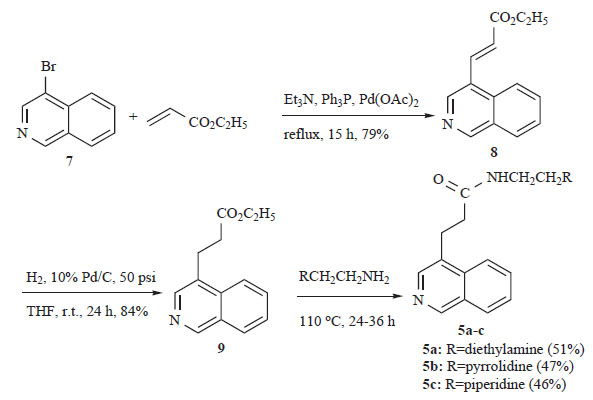
Synthesis of the new saturated amides 5a-c.

Synthesis of the new α,β-unsaturated amides 6a-c.
However, analogous treatment of the unsaturated ester 8 with the same amines did not lead to the desired compounds 6a-c in a satisfactory yield. The situation was rectified by substituting ethyl acrylate 8 with its methyl congener 10, which was prepared from 4-bromoisoquinoline (7) and methyl acrylate under Heck conditions (Scheme 2). The new 4-isoquinoline propanamides 5a-c and their unsaturated counterparts 6a-c were examined for in vitro cytotoxicity in a panel of two tumour cell lines (Table 1). In the leukemia K562 cell line none of the tested compounds showed any noticeable potency. Although most of the compounds tested were also inactive in the human non-small lung cancer cell line NSCLC-N16-L16 (an IC50 of 10 μM or lower is generally considered as “active” in anticancer drug discovery), the unsaturated analogues 6b and 6c, with IC50 values 44.0 and 35.6 μM, respectively, are considered as active in this particular tumour and suitable candidates for further biological evaluation.
Cytotoxicity of the New Analogues in the K562 and NSCLC-N16-L16 Carcinoma Cell Lines
a IC50 values were measured by using a microculture tetrazolium assay (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide, MTT) following a 72 h exposure to drug at 37°C.
b IC50 values were determined by using the MTT assay; three concentrations of the drugs were tested in duplicate and cell growth was evaluated at 72 h (Mosmann J Immunol Methods 1983) [10].
It is noteworthy that few agents, e.g. dactinomycin, are clinically effective against human non-small lung cancer, and thus there is a need for novel agents for use in this disease.
The fact that the unsaturated piperidino analogue 6c is marginally more active in this line than its congener 6b can be possibly attributed to the increased basicity of piperidine (pyrrolidine’s pka=11.11; pka of piperidine =11.29 [9]). The lack of potency in NSCLC-N16-L16 shown by the saturated analogues 5a and 5c is probably due to the different spatial orientation of their flexible side chains. Conversely, it seems that the “locked” geometry of the side chains of 6b and 6c allows them to interact in a more favorable way with the backbone of DNA (Tsotinis LDDD 2005) [11] (vide 3D structure of 6c against 5a, Fig. 2). It is interesting that the spatial disposition of the side chain of the weak saturated non-small lung cancer growth inhibitor 5b resembles that of 6c is (Fig. 2).
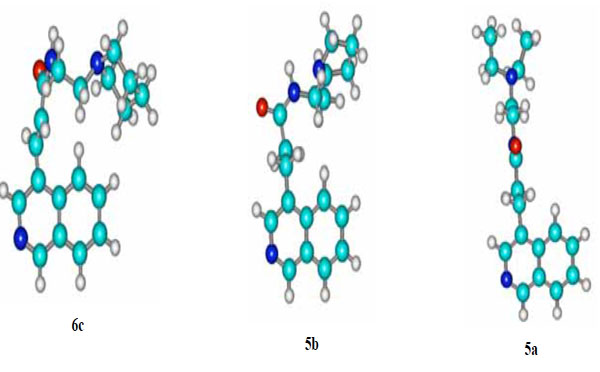
Energetically favoured conformations of 6c, 5b and 5a; the structures were obtained with the aid of HyperChem™ 7.0 Molecular Modeling System.
CONCLUSIONS
A short synthetic route to C4-substituted isoquinolines is described. Two of the analogues produced, 6b and 6c, have cytotoxicity in the NSCLC-N16-L16 cell line and may provide new leads in the search for effective cytotoxic agents.

