All published articles of this journal are available on ScienceDirect.

Chromium Coordination Compounds with Antimicrobial Activity: Synthetic Routes, Structural Characteristics, and Antibacterial Activity
Abstract
A major threat to public health worldwide is that the antimicrobial activity of the established drugs is constantly reduced due to the resistance that bacteria develop throughout the years. Some transition metal complexes show higher antibacterial activity against several bacteria compared to those of clinically used antibiotics. Novel classes of molecules provide new challenges and seem promising to solve the crisis that the overuse of antibiotics has led over the last years. This review discusses the challenges of chromium-based metallodrugs as antimicrobial agents. In particular, the synthetic routes, the structural characteristics, as well as the antimicrobial activity of 32 chromium (III) complexes have been presented.
1. INTRODUCTION
Around half of the known proteins are expected to depend on metal atoms for their structure and involvement in key cellular processes, such as electron transfer and catalysis [1]. Many metals have been used as antimicrobials since ancient times, owing to their potent toxicity to bacteria and yeast. Nowadays, antimicrobial metal compounds have many uses in manufacturing, agriculture and healthcare [2]. Single-cell prokaryotic bacteria make their home everywhere on earth, including soil, water, air, plants, and the human body [3-5]. Every year, millions of people become significantly infected with bacterial infections prompting the use of antibiotics. Due to the frequent use of antibiotics, antibiotic resistance occurs, leading to the prolonged hospital stay and increased mortality [6-8].
After 1930, a number of antibacterial medicines have been synthesized. While organic compounds have pharmaceutical activity, a primary advantage of metal-fabricated drugs over organic-fabricated drugs is their mastery to alter the number, geometry and redox states of coordination [9-13]. In addition, metals may antagonist the pharmacological properties of organic-fabricated drugs by developing compounds for coordination with them. Metal-DNA coordination complexes have been thoroughly explored over the past decades as possible anti-cancer drugs [14, 15].
In order to overcome the antibacterial resistance, different antibacterial pathways against pathogenic bacteria have been reported. It is well known that the lipid membrane covering the cell facilitates the passage of only lipid-soluble compounds. The increase of solubility of pharmaceutical compounds within the microbe membrane known as liposolubility is considered an important factor which regulates the antimicrobial activity [16, 17]. In metal-based compounds due to the partial sharing of the positive charge of metal ions with donor groups and the overlap of the ligand orbitals, the polarity of the metal ion during chelation is reduced to a great extent. In addition, the electron delocalization over the entire chelate ring is improved, enhancing the complex's lipophilicity. Once the metal-based complexes enter the cell membrane of microbes, a possible mechanism of action is by blocking the metal-binding sites of microorganism enzymes. Such metal complexes often disrupt the cell's breathing process blocking eventually the protein synthesis, which limits further organism development. The mechanism pathway of complexes against different pathogenic organisms depends either on the permeability of the microbe cell, or the difference in ribosomes of the microbe [18].
In addition, Cr (III) has been reported to interact with cysteine and histidine of cellular proteins of microbes, resulting in the formation of stable complexes. Pharmacokinetic modelling of Cr (III) accumulation, as a result of dietary supplementation, has predicted that the amount of Cr (III) linked to DNA is sufficient to reduce changes in DNA replication fidelity and increase mutation frequency [19, 20]. In addition, certain Cr (III) complexes that are soluble in physiological pH values were found to cause Salmonella Typhimurium mutagenicity. Evidence has been presented for the activation of tyrosine family kinases under the influence of Cr (III), which is, in turn, dependent on ROS development, and this may have implications for other systems that trigger apoptosis via ROS generation [21-24].
In this review paper, we present the latest Cr-coordinated compounds, which exhibit antimicrobial activity and give a critical view of how the structure can affect the action of the compound, in correlation with future perspectives of this field. In particular, 35 molecules (free ligands) are presented with their corresponding Cr (III) complexes and their antimicrobial activity against several pathogenic bacteria and the characteristics of their structure and synthetic routes are described.
2. SYNTHESIS OF THE COORDINATED COMPOUNDS
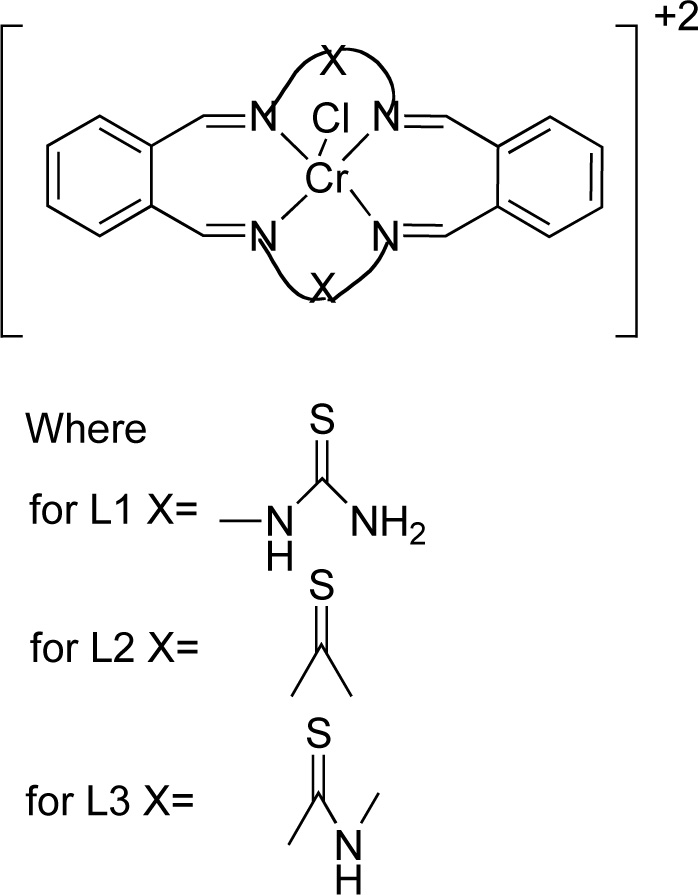
Reddy et al. [25] presented the synthesis and antimicrobial activity of six macrocyclic Cr (III) compounds. The structures of the most active ligands are shown in Fig. (1). Briefly, for the synthesis of complexes (C1, C2, C3), a methanol solution (20 mL) of ligand L1, L2 or L3 was added, at an equimolar ratio, to a chromium chloride solution (20 mL, in methanol) over stirring for 3-4 h. The resulting solution was concentrated under reduced pressure and recrystallized in diethyl ether. The precipitate was separated by suction filtration and washed with methanol and diethyl ether. The compound was vacuum dried and recrystallized again using a solvent mixture of dichloromethane and diethyl ether [Yield 70-75%].
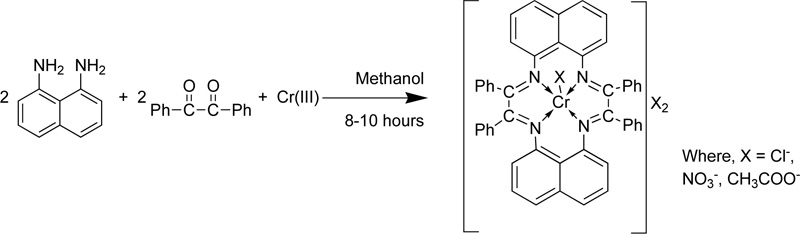
Singh et al. [26] presented the synthesis of a macrocyclic complex of Cr (III) via condensation of 1,8- diaminonaphthalence and benzil in the presence of CrCl3. Briefly, a hot methanol solution of 1,8– diaminonaphthalene (3.16% w/w) was added into a CrCl3 solution (in MeOH, ~20 mL). The resulting solution was refluxed for 0.5 h, followed by the addition of benzil (2.10 g). Then, the mixture was further refluxed for 8- 10 h. Subsequently, the mixture was condensed to half its original volume and stored overnight in a desiccator. The dark-coloured precipitates formed were filtered, washed with methanol, acetone and diethyl ether and finally dried in vacuum. The yield was 45-55%. The compound was found soluble in DMF and DMSO but insoluble in common organic and water solvents. In addition, the complex was also found thermally stable up to 260-285°C. The condensation reaction for the preparation of the complex Cr(III) with L4 (C4) is presented in Fig. (2) [27-33].
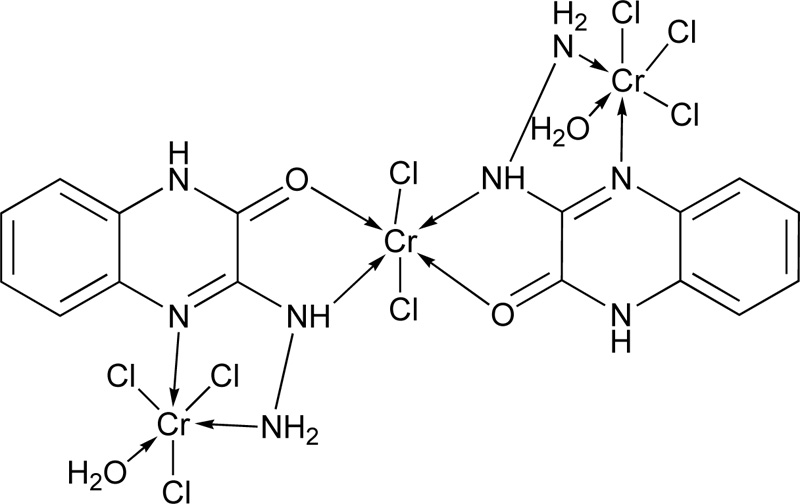
Another research group [19] presented the synthesis of 3-hydrazino quinoxaline-2-one (L5) via a two-step reaction. In the first step, the synthesis of 1,4-quinoxaline-2,3-dione was performed, followed by the synthesis of L5. The corresponding complex molecule (C5) was synthesized by the addition of a methanolic ligand solution (0.0075 moles) to a metal chloride solution (0.0025 moles) in small increments. The reaction mixture's pH was raised up to 7-8 using a methanolic ammonia solution (1% w/w), while the mixture was refluxed for 4h. The coloured product obtained was filtered and washed several times with methanol to remove any excess of the unreacted starting materials. Finally, the complex was washed with petroleum ether and diethyl ether, and dried over anhydrous CaCl2 in vacuum desiccators. The proposed structure of the complex of Cr(III) with L5 is shown in Fig. (3).
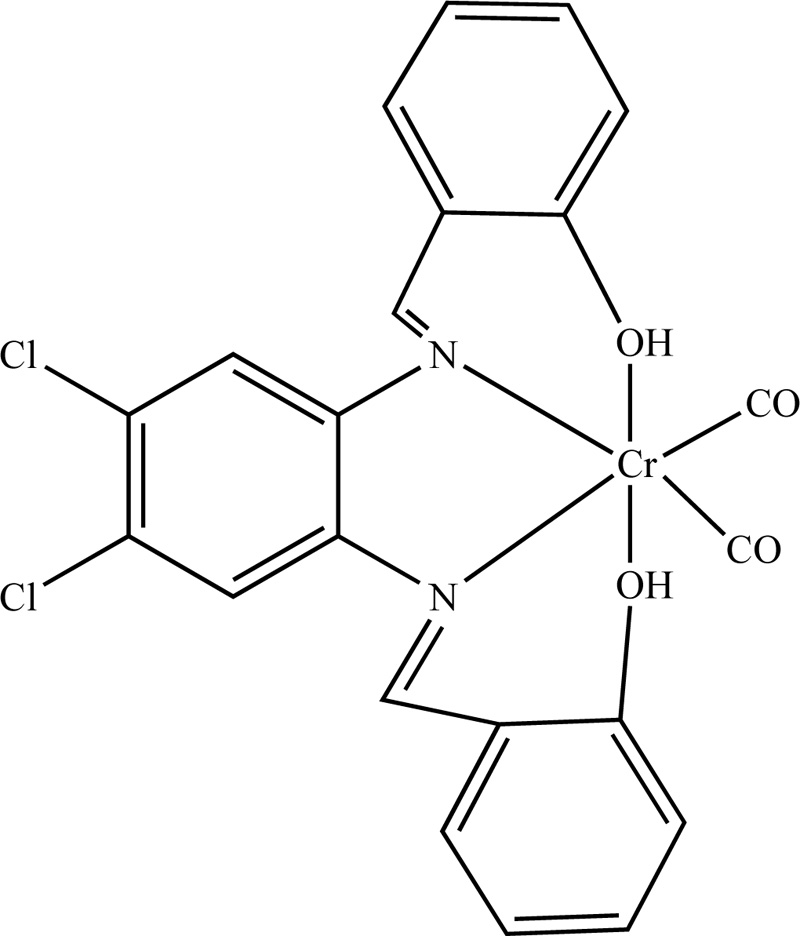
A complex of Cr(III) with N,N-bis(salicylidene)-4,5-dichloro-1,2-phenyleneediamine (L6) was also presented in literature [21]. For the synthesis of L6, 4,3 mL (40 mmol) of salicylaldehyde were added to a hot methanolic solution of 4,5-dichloro-1,2-phenyleneediamine (20 mL, 20 mmol) dropwise. The solution was then refluxed for 2h. The resulting precipitate Schiff base was isolated by filtration and recrystallized from methanol. The ligand dried in a desiccator over anhydrous CaCl2 under vacuum. The resulting product was washed with ethanol and received at a yield of 63%. Subsequently, the synthesis of Cr(III) with L6 (C6) complex was accomplished by the addition of a ligand solution in dry benzene (0.45 mmol, 20 mL benzene) into the salt, Cr(CO)6 (0.45 mmol). The resulting blend was degassed and heated at 80°C for 5 h. Then, the reaction mixture was cooled, followed by the evaporation of the solvent under vacuum. The brown residue was washed with hot petroleum ether several times and recrystallized. (Yield 74%) The proposed organized compound structure is illustrated in Fig. (4).
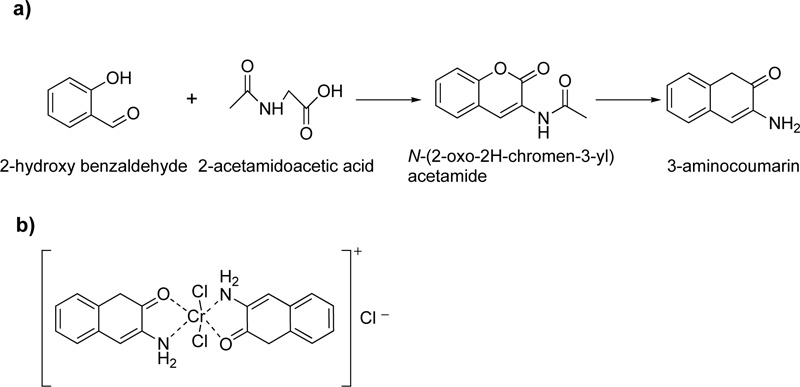
Moreover, a Cr (III) coordinated compound derived from 3-aminocoumarin ligand, L7, is described in literature.
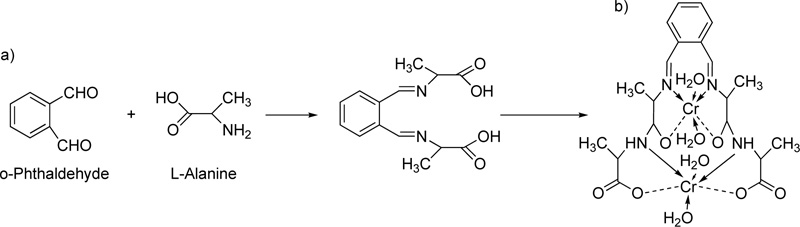
[11] In the presence of piperidine as a strong base, 2-hydroxybenzaldehyde and 2-acetamidoacetic acid reacted by vigorous reflux, resulting in the formation of L7. The resulted octahedral complex of Cr(III) with L7 (C7) is shown in Fig. (5a and b).
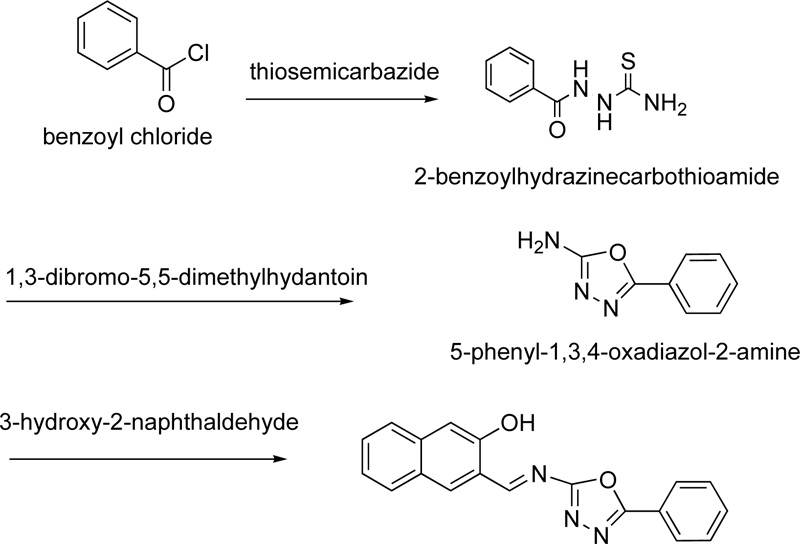
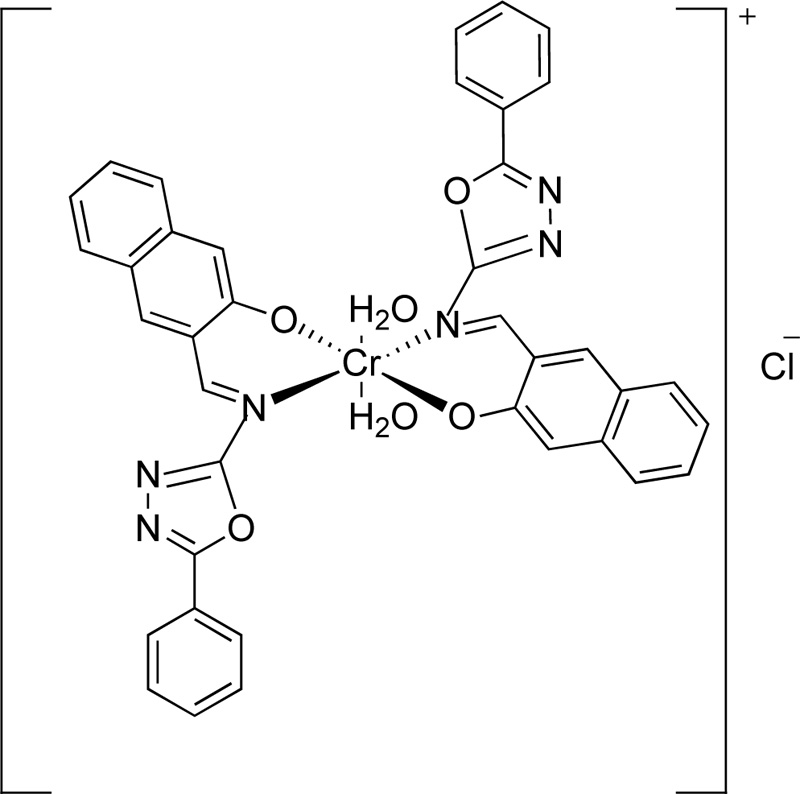
A year later, the synthesis of (E)-3-(5-phenyl-1,3,4-oxadiazol-2-ylimino)methyl)naphthalene-2-ol, L8, as a ligand for Cr(III), was reported by Amiery [31]. For the synthesis of the ligand, equimolar amounts of 3-hydroxy-2- naphthaldehyde and 2-amino-5-phenyl-1,3,4-oxadiazole were refluxed in ethanol (in the minimum required amount) for 2 h. After the cooling of the mixture, it was separated and washed with distilled water, ether and cold ethanol several times. Finally, the ligand was dried and recrystallized from acetone giving yellow needle-shaped crystals. (Yield 87%) The synthesis of complex (C8) was performed, first, by mixing a hot ethanolic solution (50 mL, 2 mmol) of L8 with an ethanolic solution of CrCl3.6H2O (20 mL, 1 mmol), followed by the addition of 10% w/v sodium acetate until the complex precipitation (pH 8.5). The complex was filtered, washed with water, ethanol and dried under vacuum. The synthetic route followed for the preparation of L8 is indicated in Fig. (6), while Fig. (7) presents the proposed structure of Cr(III)-L8 complex.
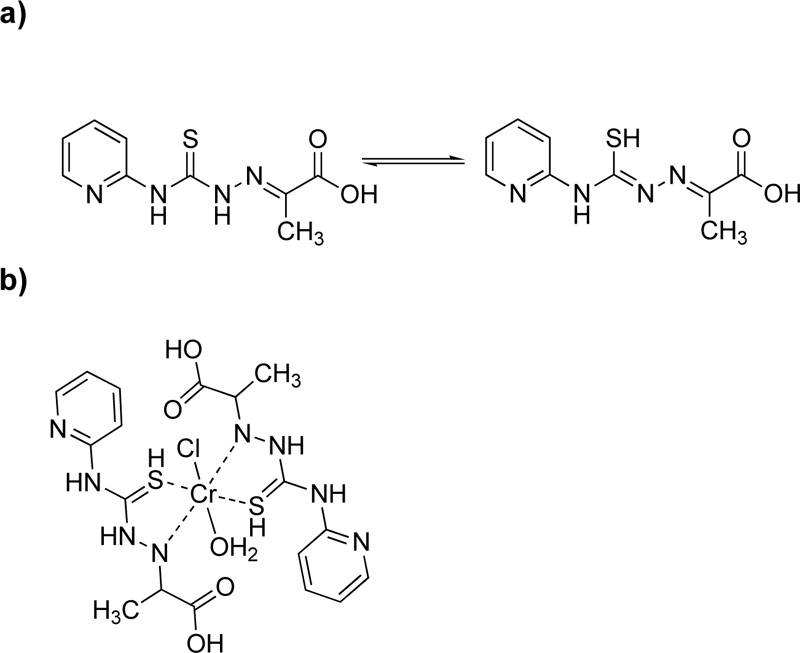
Moreover, a new ligand, L9, was prepared via the condensation reaction of 4-(2-pyridyl)-3-thiosemicarbazide and pyruvic acid at a molar ratio of 1: 1 Fig. (8a) [24]. For the purification, the mixture was filtered, washed with ethanol and recrystallized in ethanol. (Yield 89.6%) The metal complex (C9), Fig. (8b), was synthesized via the addition of chromium chloride (10 mmol) into a hot ethanol solution of L9 (4.76% w/w, 10 mmol). It is worth mention that L9 exhibits thiol-thione tautomerism in which sulfur plays an important coordinating role [34].
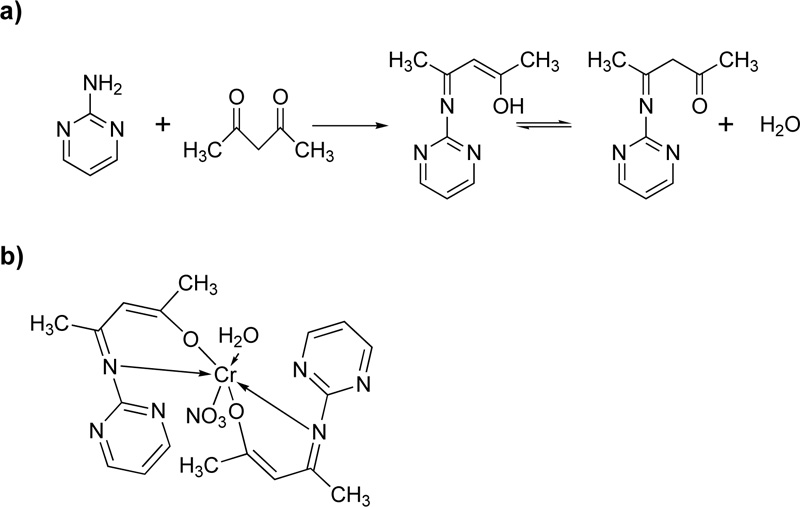
Furthermore, Oruma et al. [18] presented the synthesis of (2Z,4Z)-4-(pyrimidin-2-ylimino) pent-2-en-2-ol (L10) ligand Fig. (9a). For the preparation of L13, an ethanolic solution of 2-aminopyrimidine (0.02 mol) was added to an ethanolic solution of pentan-2,4-dione (0.01 mol) and refluxed for 6 h. The resulting pale-yellow solution was cooled, and recrystallized in ethanol.
For the synthesis of the Cr(III)-L10 complex (C10), an ethanolic solution (50 mL) of Cr(NO3)3.H2O (0.002 mol) was added to L10 (0.04 mol). The mixture was refluxed for 2h, condensed to half its volume, and placed over anhydrous CaCl2 in a desiccator for 2 days. The green precipitate formed was filtered off, washed with ethanol and dried. The proposed complex structure is shown in Fig. (9b).
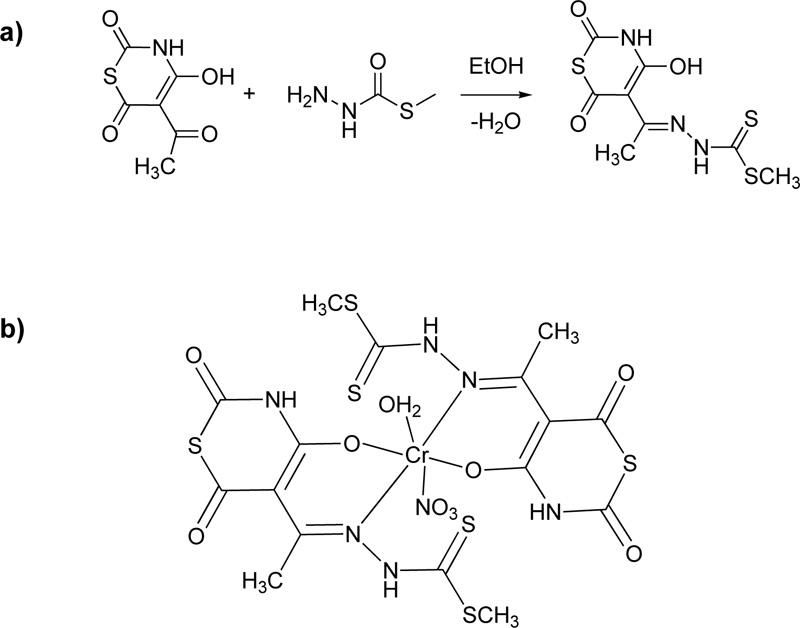
Another ligand (L11, Fig. (10) for Cr(III) coordination was proposed by Taha et al. [36] For the preparation of this ligand, an ethanolic solution of 5-acetyl-4-hydroxy-2H-1,3-thiazine-2,6(3H)-dione (18.7% w/w) was added into a hot ethanolic solution of 5-methyl dithiocarbazate (12.2% w/w) and the reaction mixture was refluxed for 1 h. The yellow crystals received after cooling were filtered, washed several times with ethanol and diethyl ether and dried over solid CaCl2 in a desiccator. Finally, fine crystals were obtained by recrystallization from ethanol. (Yield 55%). Subsequently, a deprotonated solution of the L11 (2.92% w/w) which was prepared in the presence of LiOH (ratio L11: LiOH, 1: 2) was added to a methanolic solution of the metal salt (Cr(NO3)3, 3.38% w/w) in order to receive the complex (C11). Fig. (10a) and (10b) illustrates the synthesis of L11 and the proposed structure of the coordinated compound, respectively.
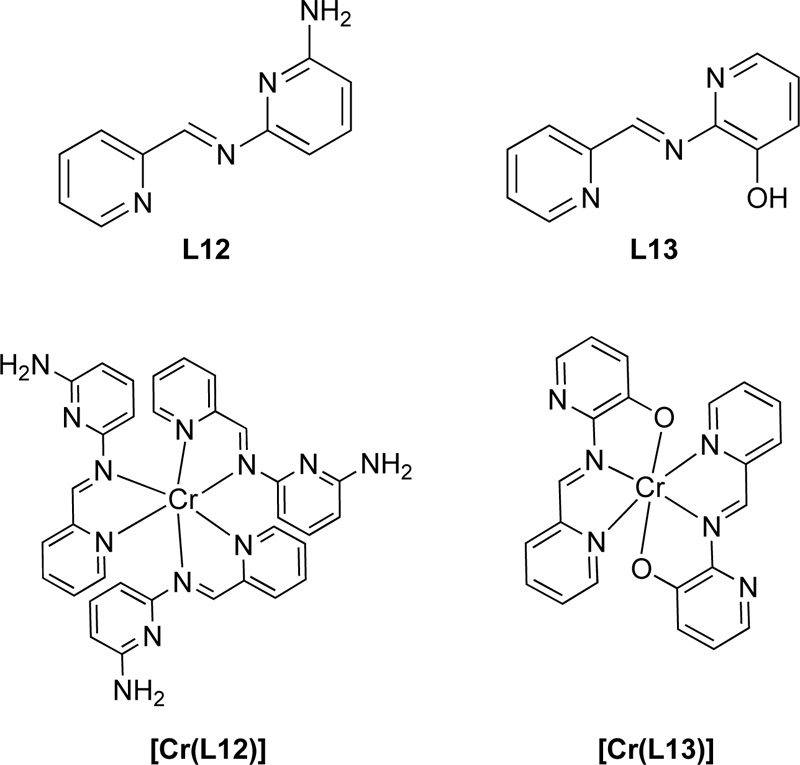
Moreover, another research group [35] proposed the synthesis of two Schiff base ligands, the 2-[(pyridin-2- ylmethylidene)amino]-6-aminopyridine (L12) and the 2-[(pyridin-2-ylmethylidene)amino]phenol (L13). For the preparation of L12, a solution of 2-pyridinecarboxaldehyde (9.33 mmol) in ethanol was added to 1.018 g of 2,6- diaminopyridine (9.33 mmol) and the mixture was refluxed for 15 h. The resulting solution was then concentrated by evaporation, filtered and washed with ethanol several times resulting in a solid brown product. Ligand was finally recrystallized in ethanol. (Yield 88%) For the synthesis of L13, a methanolic solution of 2- pyridinecarboxaldehyde (8.4 mmol) was added to a methanolic solution of 2-aminophenol (8.4 mmol) and the solution refluxed for 11 h. The resulting solution was purified following the same procedure as L15 giving a yield of 78%. Chemical structures of both L12 and L13 are presented in Fig. (11).
Regarding the preparation of Cr (III) complexes (C12, C13) with the L12 and L13, a mixture of equimolar amounts of [Cr(CO)6] and L15 or L16 in DMF: THF (ratio of 1: 2) solvent mixture (30 mL) was heated to reflux. Subsequently, the solvent was removed on a vacuum line and the residue was washed several times with boiling petroleum ether and, finally, recrystallized in DMF. Fig. (11) represents the complexes of Cr(III) with L12 and L13.
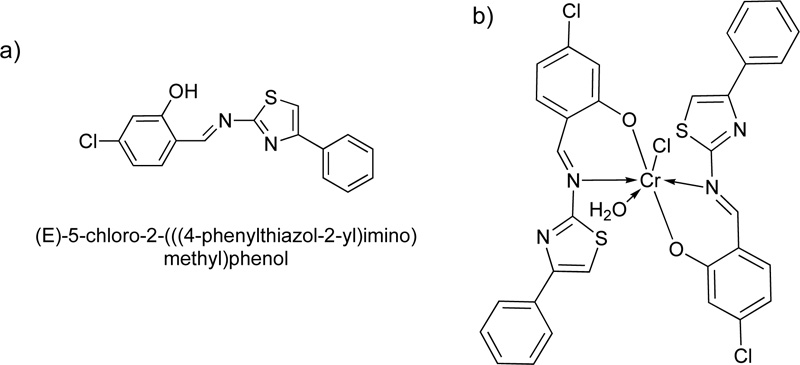
A year later, a new Cr(III) complex with 5-chloro-2-4-phenylthiazol-2-yl-iminomethylphenol (L14) ligand was reported [36]. For the preparation of the Schiff base L14, an ethanolic solution of 4-chloro-2- hydroxybenzaldehyde (24.88 mol) was mixed with 4-phenylthiazol-2-amine (24.88 mmol) and refluxed for 15h. The resulting solution was then condensed by evaporation, filtered and washed with ethanol several times. The crystals received were further suction filtered, washed with diethyl ether and dried. The purity of the product was checked by TLC (Yield 88%, melting point 138°C).
After the purification of the ligand, the Cr (III) complex (C14) was prepared by adding a solution of chromium chloride hexahydrate, CrCl3.6H2O, (6.65% w/w in ethanol) into a solution of L14 (1.) dropwise. The reaction mixture was stirred for 3 h at room temperature. The apple-green precipitate was filtered off and dried under vacuum over CaCl2. The chemical structure of the ligand, as well as its complex with Cr(III), are illustrated in Fig. (12a and 12b), respectively [37].
The next work [38] describes the synthesis of a novel bidentate Schiff base (L15)via the condensation reaction of 2-amino-4-phenylthiazone with 4,6-diacetylresorcinol at a molar ratio of 2:1 Fig. (13). Briefly, the ligand was synthesized by adding an ethanoic solution (12.9% w/w) of 4,6-diacetylresorcinol to an ethanoic solution (23.5% w/w) of 2-amino-4-phenylthiazole. The reaction mixture was heated in reflux for 4 h, while the yellow crystals received were filtered and washed off with ethanol and ether, and dried in air. Finally, fine crystals were obtained by recrystallization in ethanol (Melting point: 212-213°C, yield: 78.2%). Subsequently, 2.55 g of L15 (5.0 mmol) and 2.38 g of metal salt (Cr(NO3)3, 10 mmol) were dissolved in 50 mL of ethanol, stirred for 30 min (at RT) and then heated over reflux for 3 h. The solvent of the mixture was removed via evaporation, while the product was filtered off, washed with ethanol and diethyl ether and finally dried. The complex (C15) received was soluble in most common solvents.
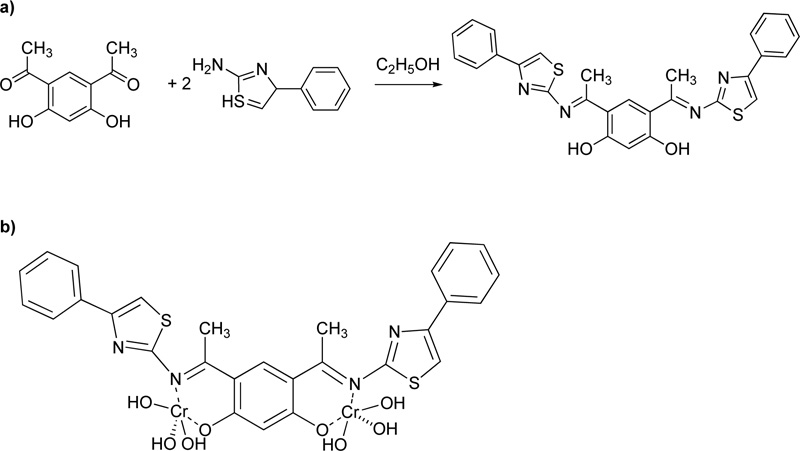
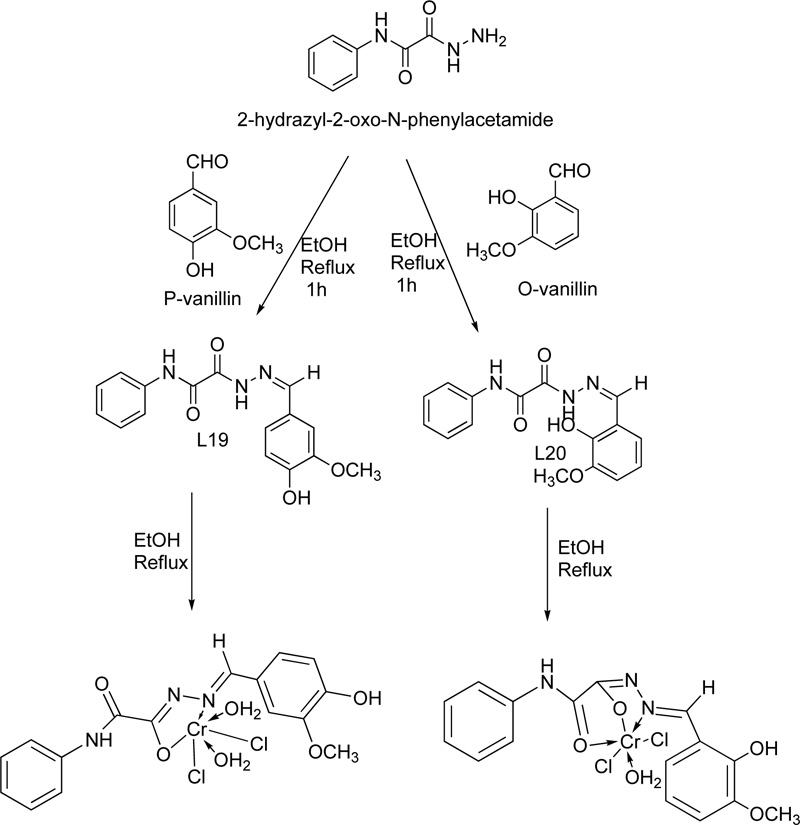
Two Cr(III) complexes with hydrazone ligand are also described in the literature [39]. The two ligands (L16, L17) were prepared via the condensation reaction of 2-hydrazyl-2-oxo-N-phenylacetamide with p-vanillin or o- vanillin at a molar ratio of 1: 1. The reaction performed over reflux for 1 h. For the purification, the precipitates were filtered, washed several times with ethanol and finally dried in a vacuum desiccator over anhydrous CaCl2. The synthesis of the complexes took place via the addition of a hot ethanolic solution of the respective metal chloride (1.0 mmol) to a hot ethanolic solution of the ligand (1.0 mmol). The resultant mixture was heated under reflux for 3-4 h. The complexes were filtered off, washed with either hot ethanol (L16) or diethyl ether (L17) and dried in a vacuum desiccator over anhydrous CaCl2. The synthetic procedure followed for the synthesis of the Cr(III) with L16 (C16) and Cr(III) with L17 (C17) complexes is shown in the figure below Fig. (14).
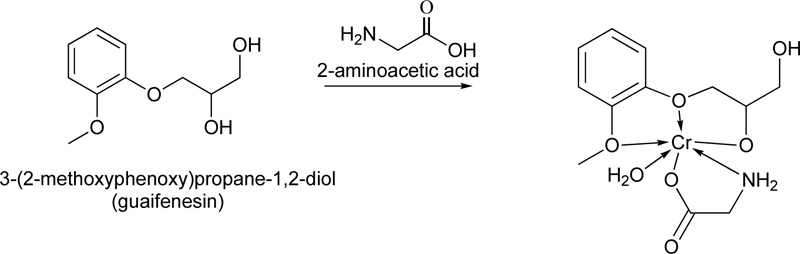
In addition, a very interesting complex using guaifenesin (L18) as ligand has been reported as well [40]. Briefly, the metal complex (C18) was prepared by mixing equal quantities (0.02 mol) of the guaifenesin (hot saturated ethanolic solution) and metal chloride (CrCl3.6H2O) in the presence of 2-aminoacetic acid (0.02 mol) Fig. (15). The mixture was reflux for 3 h, followed by its purification via filtration, washing with hot ethanol and drying. (Yield ranged: 60% - 94%)
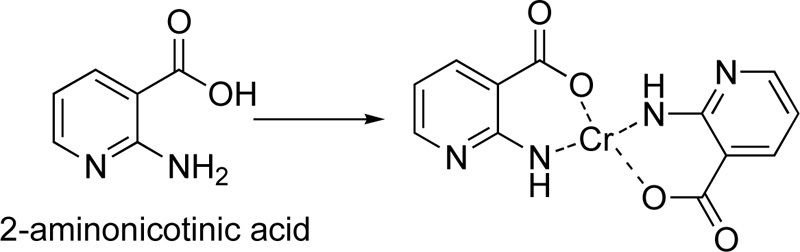
A complex of Cr(III) (C19) with 2-aminonicotinic acid (L19) as a ligand was presented by Nawaz et al. [41] For the preparation of the complex, 0.28 g (2 mmol) of 2-aminonicotinic acid and 0.11 g (2 mmol) of potassium hydroxide were mixture and stirred in deionized water for 30 min, followed the addition of an aqueous solution of metal salt (0.5 mmol). The mixture was further stirring for 30 min. The precipitate formed was filtered, washed several times with water, dried and stored in silica gel desiccator. The proposed structure of the complex molecule with L19 is depicted in Fig. (16).
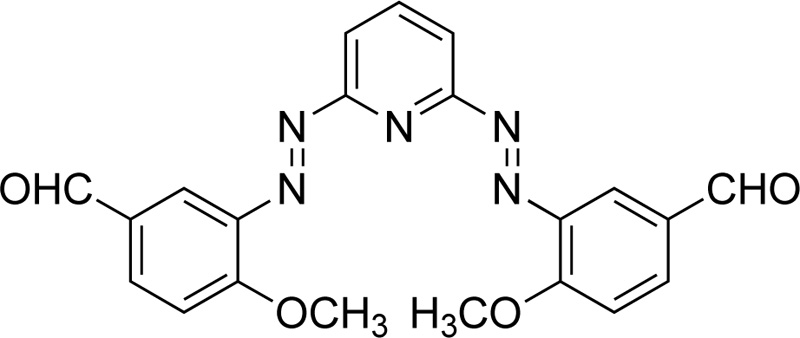
Furthermore, Mahmoud et al. [42] proposed the synthesis of an azo dye ligand (L20). For the synthesis of L20, firstly, 2.6-diaminopyridine was dissolved in 50 mL of ethanol (6% w/w), followed by the addition of a solution of HCl (4 ml) at 0ºC. Subsequently, a solution of sodium nitrite in water (20 mL, 18.8% w/w) was added dropwise and the resulting mixture was then transferred to an ethanolic solution (30 mL) made of p- methoxybenzaldehyde (24.7% w/w) and sodium acetate (13,3% w/w) and left for stirring for 1h at 0ºC. The sodium acetate served as a catalyst for the reaction. The product was collected by filtration, washed several times with water and finally dried under vacuum at RT. (Yield 85%, red solid). Subsequently, a solution of metal salt (CrCl3.6H2O, 0.993 mol) in ethanol was added to a solution of L20 (in DMF, 0.993 mmol). The mixture was heated under reflux for 2-3 h. The precipitates received were filtered, washed with ethanol and diethyl ether and dried in a vacuum desiccator over anhydrous CaCl2. Fig. (17) presents L20.
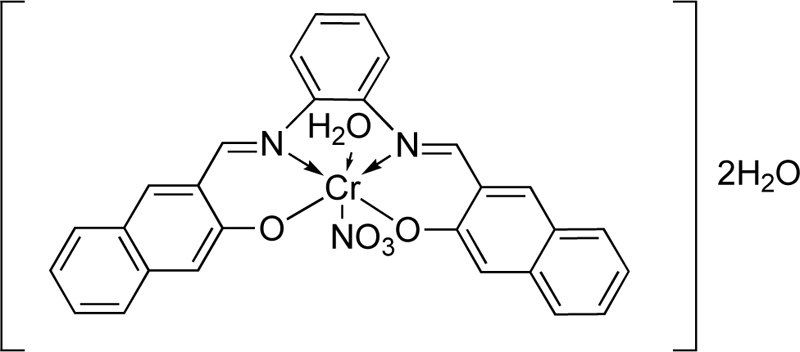
A novel tetra-dentate imine ligand (L21) was also coordinated to Cr(III) [28]. The synthesis of L21 was performed via the condensation o-phenylenediamine (5 mmol) with 2-hydroxy-1-naphtaldehyde (10 mmol) in ethanol. The resulting mixture was refluxed at 80°C for 1h, while the completion of the reaction was monitored by TLC. The obtained orange and yellow crystals were filtered, washed with cold ethanol several times and dried under reduced pressure in a desiccator. Cr (III) complex was (C21) synthesized by the addition of a solution of Cr(NO3)3.6H2O (30 ml in ethanol, 13.3% w/w) into a hot ethanolic solution (30 ml) of L24 (13.9% w/w). The resulting mixture was refluxed for 1 h. Finally, after the evaporation of solvent, the crystals were washed vigorously with ethanol and desiccated over anhydrous CaCl2. The complex molecule of Cr (III) with L24 is indicated in Fig. (18).
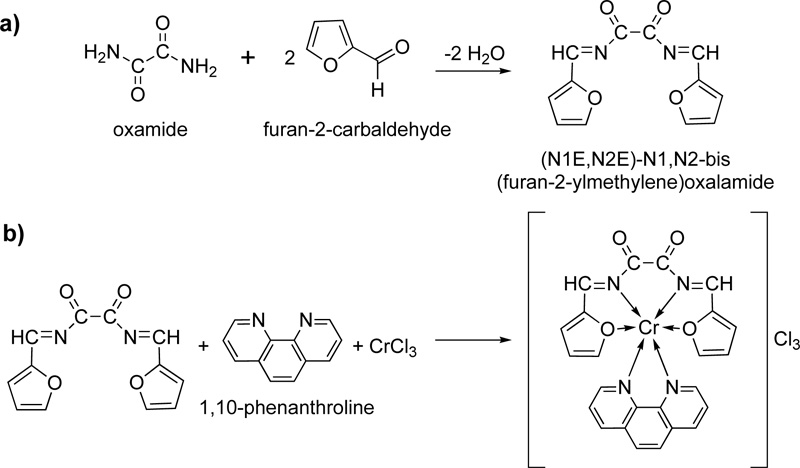
Another new Schiff base (L22) was prepared and coordinated to Cr (III) [44]. The synthesis was accomplished via the condensation reaction of furan-2-carbaldehyde and oxamide. Briefly, 22.72 mmol of furan-2- carbaldehyde (dissolved in hot ethanol) and 11.36 mmol of oxamide (dissolved in hot DMF:ethanol mixture 3:1) were left to react under reflux for 3h. The received pale brown product was separated by filtration, washed with ethanol and dried under vacuum over anhydrous CaCl2. The recrystallization in ethanol resulted in a pure Schiff base with a yield of 75%. The complexation took place by mixing a solution of L22 (20 ml, 2.5% w/w in DMF) as a primary ligand, a solution of 1,10-phenanthrolinec (20 mL, 2.3% w/w in ethanol) which served as secondary ligand and a solution of the metal salt (20 mL, 2.65% w/w in absolute ethanol). The resulting mixture was reflux for 1h, followed by its purification via filtration and recrystallization. The reactions for the synthesis of L22, as well as its complex with Cr(III) (C22), are shown in Fig. (19a and 19b), respectively.
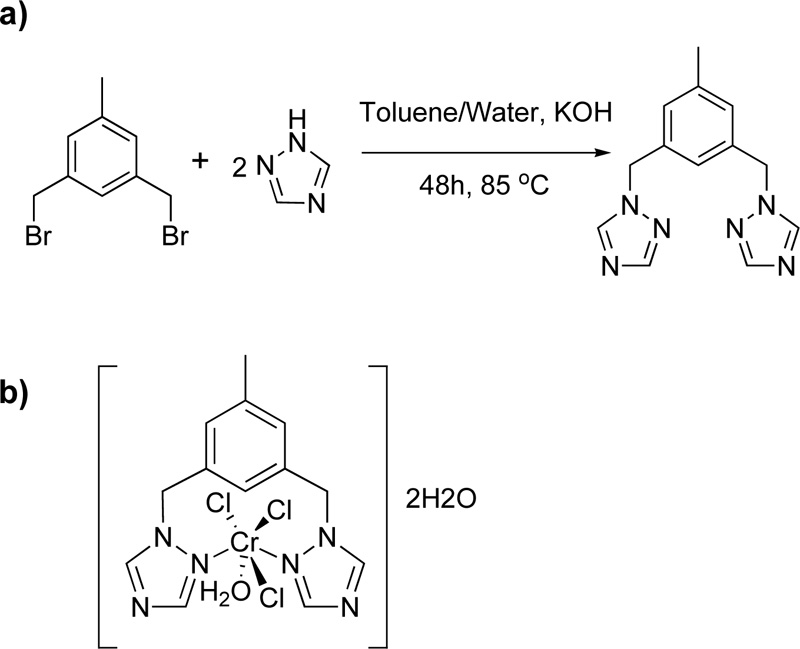
Furthermore, Murcia et al. (102) introduced the synthesis of a triazole ligand, named 3,5-bis(1,2,4-triazol-1- ylmethyl)toluene (L23), via a phase-transfer catalysed reaction of 1,3-bisbromomethyl toluene with 1H-1,2,4- triazole in the presence of THF and water as solvents. (Yield 67%) A solution of chromium salt in acetone (CrCl3.6H2O, 2.21% w/w) was then added to a solution of L23 in acetone (4.03% w/w) and the mixture was stirred for 2 h in RT. The mixture was centrifuged for 8 min at 400 rpm, washed with acetone, DCM and diethyl ether. The resulting green complex (C23) was air-stable and non-hydroscopic. The reaction for the synthesis of L23 and the structure of its complex with Cr (III) is depicted in Fig. (20a) and Fig. (20b), respectively [45, 46].
Another interesting amino acid Schiff base (L24) was reported by Babu and co-workers [47]. For the ligand synthesis, first, a solution of alanine (0.58 g), L-prenylalanine (3.72 g), L-lycine (3.72 g), methyldopa (4.22 g), glutamate (3.38 g), L- tryptophan (4.8 g), L-histidine (3.1 g) and glycyl-glycine (0.64 g) in methanol (10-30 mL) was prepared, followed by the addition of an alcoholic solution of KOH. The solution was stirred for 1h and then filtered. Subsequently, a solution of o-phthalaldehyde (6.6% w/w, in methanol) was added dropwise. The product was evaporated and washed with cold ethanol and diethyl ether and finally dried. The complex (C24) synthesized by the addition of the chromium salt (0.005 mol) to an L24 solution in hot methanol (0.005 mol, 25 ml), and the mixture was refluxed for 2 h until a solid was separated. The solids were filtered to suction, washed with CHCl3, methanol and dried with anhydrous CaCl2 over the vacuum. Fig. (21) shows the synthetic route for the preparation of L24 Fig. (21a as well as its complex with Cr. Fig. 21b) [48, 49].
Moreover, the synthesis of 2-hyroxybenzaldehyde-4-allyl-S-methylsothiosemicarbazone (L25) has been proposed [17]. Briefly, for the synthesis of L25, 4-allyl-3-thiosemicarbazide (1.31 g) was dissolved in 20 ml of ethanol, followed by the addition of iodomethane (1.56 g). The mixture was stirred at RT for 2 h, followed by the addition of 2-hydroxybenzaldehyde (1.22 g). The mixture was stirred for 30 min at 80°C. After the reaction mixture was cooled, the yellow precipitate which was formed was filtered, washed with ethanol and dried in vacuum. The complex (C25) was synthesized by the addition of Cr(NO3)3.9H2O (1mmol) in a solution of L25 (2 mmol, in 60 mL ethanol), while the mixture was refluxed for 1 h.
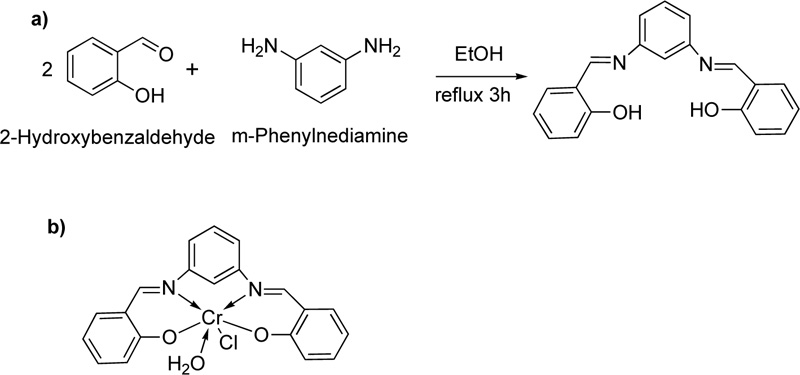
Recently, another ligand (L26) has been introduced [50]. L26 was prepared by the condensation reaction of m- phenylenediamine with 2-hydroxybenzaldehyde Fig. (22a). In particular, a solution of 2- hydroxybenzaldehyde (74 mmol) dissolved in ethanol was added dropwise to a solution of m-phenylenediamine (37 mmol, in ethanol). The resulting mixture was stirred under reflux for about 3 h at 100-150°C, where an orange precipitate was separated. The latter was filtered, recrystallized with diethyl ether and dried in vacuum. (Yield 53%) Subsequently, a solution of L26 (0.4 g) in a mixture of ethanol and DMF (Ethanol: DMF, 2: 1, at 60°C) was added to a solution of chromium chloride (1.266 g, in ethanol, 60°C). The resulting mixture was stirred under reflux for 2 h where the complex (C26) was precipitated. Crystals were collected by filtration, washed with hot ethanol and purified by washing several times with diethyl ether.
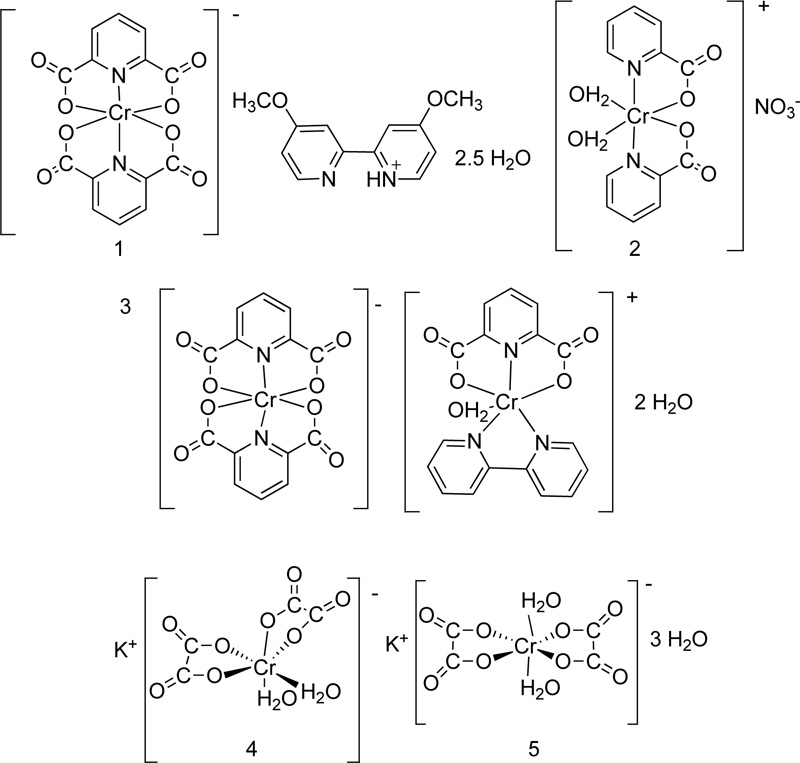
Five more Cr (III) complexes were prepared and tested for their antimicrobial activity [51, 52]. In particular, the following ligands picolinate (L27), dipicolinate (L28), oxalate (L29), 2,2-bipyridine (L30) and 4,4-dimethoxy- 2,2-bipyridine (L31) were coordinated to Cr(III) Fig. (22). The synthesis of complexes (C27, C28, C29, C30, C31) was performed via the reaction of the metal chloride salt (CoCl3.6H2O) with the corresponding ligand, in a molar ratio of 1:1, in the presence of ethanol as solvent. The reaction proceeded for 3-4 h in reflux, while the precipitates were washed with cold ethanol and dried over CaCl2 in a vacuum. All the proposed structures of the complexes prepared are illustrated in Fig. (23).
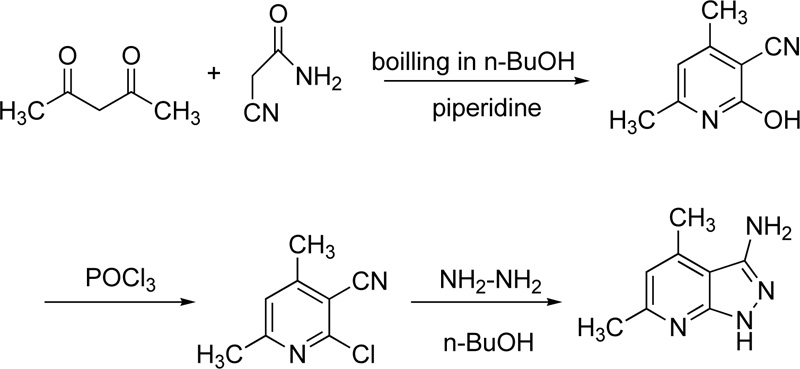
Finally, very recently, Masoud et al. reported the chelating behaviour of 3-amino-4,6-dimethyl-1H-pyrazolo [3,4-b] pyridine (L32) towards several metal ions, including Cr(III) [53]. Briefly, the synthesis of L35 was performed via three steps. Firstly, the synthesis of 2-hydroxy-2,6-dimethylpyridine-3-carbonitrile took place via the reaction of cyanoacetamide with acetylacetone in boiling n-butanol in the presence of piperidine (few drops). In a second step, 2-hydroxy-2,6-dimethylpyridine-3-carbonitrile reacted with phosphorus oxychloride (POCl3) to give 2-chloro-4,6-dimethylpyridine-3-carbonitrile. The latter compound reacted subsequently with hydrazine hydrate in boiled n-butanol to give 3-amino-4,6-dimethyl-1H-pyrazolo [3,4-b] pyridine Fig. (24).
The metal complex (C32) was prepared by adding the metal chloride salt into an ethanolic suspension of L32 at a molar ratio metal salt: L32 of 1: 2. The reaction mixture was refluxed. The resulting mixture was cooled and the precipitate was filtered, washed several times with ethanol, and dried over anhydrous CaCl2 in a vacuum desiccator.
3. STRUCTURAL CHARACTERISTICS OF THE COMPOUNDS
The synthetic route followed for the synthesis of 32 ligands (L1-L32), as well as the preparation of their corresponding complexes with Cr(III), has been described above. In this section, the structural characteristic of all complexes which confirmed their successful formation, will be reviewed.
The confirmation of C1, C2 and C3 was confirmed first via the use of Infrared (IR) spectral data . For C1, a medium intensity band due to C=N bond was shifted towards the lower side, about 20-30 cm-1, compared to the ligand spectra and appeared in the range of 1625-1600 cm-1. The same observation was also observed for C2 and C3. These findings are further confirmed by the presence of a band assignable to metal-nitrogen vibration within the 525-505 cm-1 zone. The electronic spectra of C1, C2 and C3 show bands at 17450-18340 cm-1, 27440-27820 cm-1 and 34830 cm-1, which were consistent with those predicted using X-ray measurements [25]. Regarding the C4 molecule, a pair of bands presented in the IR spectrum of 1,8-diaminonaphthalene at 3350 and 3390 cm-1 corresponding to the NH2 group, which were absent in the IR spectrum of the C4, confirming its successful formation [52, 54, 55]. A lower value of C=N stretching, which was also appeared in the IR spectrum, was attributed to the drift of a lone pair density of azomethine nitrogen towards the metal atom [64, 65], indicating that coordination takes place through the nitrogen of (C=N) groups. Magnetic moments of C4 were found in the range of 4.15-4.52 B.M. at RT, which is close to the predicted values for the three unpaired electrons presented in the metal ion. The electronic spectra of C4 show bands at 9010-9320 cm-1, 13030- 13340 cm-1, 17460-18320 cm-1, 27420-27850 cm-1 and 34810 cm-1. The spectral bands are consistent with that of the five coordinated Cr (III) complexes, whose structure has been confirmed with the help of X-ray measurements [55, 57, 58]. The latter led to the assignment of a five coordinated square pyramidal geometry to C4. IR spectra of C5 showed broad peaks in the region of 3660-2850 cm-1, which were attributed to v(OH) of the coordinated water as confirmed by thermal analysis as well. The rocking mode of coordinated water is also observed in the range of 900-910 cm-1. It is assumed that the v(NH of hydrazine), v(ring NH) are merged with v(OH) of coordinated water. The NMR spectra of C5, indicated a preference for amide form in metal binding [59-76. This was distinctly observed from the absence of OH signal at δ10.1 Coordination through amide oxygen was established by the negative shift in v(C=O), as shown by the IR spectra. The -NH2 protons of hydrazine undergo a significant downfield shift from δ 4.7 to δ 7.5-7.7, which further confirmed metal-binding through the corresponding nitrogen [77, 78]. C5's electronic spectra displayed many bands that can be clarified by imagining different conditions around each metal ion. The abundance of electronic spectral bands within this complex also provided proof of the complex's trinuclearity.[19, 70]. Based on these data, octahedral geometry was proposed for C5. For the C6 complex, its IR spectrum showed that the characteristic band of the azomethine nitrogen atom of the free L6 at 1616 cm-1 was shifted to the lower wavenumber region (1599-1610 cm-1) after complexation, indicating the involvement of the azomethine nitrogen atom in coordination [68, 69]. Another evidence of the bonding was observed in the IR spectrum of C6, where two new peaks appeared at 451-467 cm-1 and 524-530 cm-1 were assigned to (M-N) and (M-O) stretching vibrations. In addition, a strong band due to the Cr=O at 851 cm-1 was also exhibited. Furthermore, the 1HNMR spectrum of the parent Schiff base L6 showed a singlet signal at 12.47 attributed to the two phenolic -OH protons. L6 also showed one singlet at 8.89 ppm, which attributed to the two azomethines (-CH≡N-) protons. The proton NMR spectrum of the L6 revealed a multiplet at 6.87-7.74 ppm corresponding to aromatic protons. Coordination of both ligand nitrogen atoms to the metal centre was confirmed by a signal shift from 8.89 ppm for the free ligand to 8.72 and 8.50, respectively, for the imine carbon protons. The 11.67 ppm signal suggested that -OH groups were present. The signal showed a lower field shift and indicated that the ligand coordinated to the Cr atom through the OH group. The C6 diamagnetism indicated the presence of Cr in a zero oxidation state with a low electronic spin d6 configuration [21].
A study of the IR spectra of L7 and C7 implied that L7 was bidentate with the carbonyl oxygen and amine nitrogen as the two coordination sites. In the IR spectrum of C7, a considerable negative shift of the C=O bond as a consequence of coordination through the carbonyl oxygen atom was detected. The carbonyl band of the parent L7 was located at 1709 cm-1, while that of the C7 was shifted to the lower frequency at 1691 cm-1. The NH2 bands of the ligand were found at 3407 and 3299 cm-1, but those of the complex was shifted to lower frequencies, at 3315 and 3201 cm-1, respectively. Additional evidence for the coupling between the ligand and metals ion was the presence of M-N and M-O peaks at 455-467 cm-1 and 486-503 cm-1, respectively. The C7 molar conductivity value showed one chloride ion outside the sphere of coordination. C7 was viewed as an octahedral structure [11]. L8 showed two moderately strong bands at 3400 and 1630 cm-1 assigned to O-H and C=N groups. The absorption band in the range of 460-515 and 380-430 cm-1 were assigned to M-N and M-O bands. The molar conductance of C8 indicated that the chloride ion was present outside the octahedral coordination sphere. The ultraviolet (UV) spectrum of L8 showed two absorption bands, the first band at 240 nm corresponded to a π→π* transition, while the second band appeared at 315 nm attributed to n→π* transition. All the UV bands of C8 were shifted to a lower wavelength and had higher intensity. The C8 complex showed a magnetic moment of 3.2 B.M, which supported the high spin octahedral geometry [31]. For C9, the absence of the peak of v(S-H) in the IR spectrum of C9 suggested the coordination of sulfur through deprotonation. The thioamide band, which contained considerable v(CS) character, was less intense in the complex and found at a lower frequency, suggesting coordination of the metal through sulfur after deprotonation [79]. The lower shift of v(C=N) stretching vibration of the azomethine in the C9 indicated the participation of the azomethine nitrogen in coordination [66, 67]. The pyridine in-plane deformation mode at 614 cm-1 in the spectrum of the ligand, shifted to 635-645 cm-1 IR spectrum of C9, suggesting coordination of the heteroaromatic nitrogen. The electronic spectra of C9, showed two strong bands at 16,989 cm-1 (u1) and 19,689 cm-1 (u2). The ligand field parameters 10Dq (1918.9 cm-1) and B (783 cm-1) were an additional evidence for the octahedral geometry [24]. For C10, the important IR vibrational frequencies were at 3482 cm-1 v(O-H), 1630 cm-1 v(C=N), 1378 cm-1 v(C- O), 613 cm-1 v(M-O) and 550 cm-1 v(M-N). The relevant electronic spectra data of C10, showed two bands at 13713 and 11198 cm-1, respectively, assigned to d-d transitions. For the C11, the absence in the IR spectrum of both v(C= S) and v(NH) indicated thioenolization of the C= S group and attachment of the thiolic sulfur to the metal ion. The latter was further confirmed by the presence of a new absorption band in the range of 671-680 cm-1 ascribed to the v(C-S). A strong band due to the v(C=N) appeared at 1645 cm-1 in the free ligand was shifted by 49-53 cm-1 in the C11 molecule, indicating the participation of the azomethine in complex formation. The bathochromic shift of v(N-N) band of C11 in the range of 1116-1079 cm-1 compared to the v(N-N) band in the free ligand at 1038 cm-1, was another clear evidence for the coordination via thiol and not thione sulfur. The unidentate nature of NO3− anion in the C11 was confirmed by the appearance of three characteristic bands at 1384, 1329 and 827 cm-1 for a monodentate nitrate group. The electronic spectrum of the green C11 showed two absorption bands at 16366.6 cm-1 and 24630.0 cm-1, suggesting an octahedral geometry [36]. Regarding C12, its two NH2 stretching modes were observed at 3332-3348 cm-1 and 3197-3280 cm-1 for the asymmetric and symmetric stretching vibrations, respectively. The in-plane ring deformation bands of pyridyl and azomethine C=N of C12 were shifted to higher wavenumbers, suggesting coordination between pyridyl and azomethine nitrogen atoms [96]. A non-ligand band at 404 cm-1 due to Cr-N bond was also indicated. Therefore, Cr atom most probably was coordinated to L12 through the azomethine nitrogen and pyridyl nitrogen. Magnetic studies of C12 showed paramagnetic characteristics. The magnetic measurements at 298 K gave a value of 2.98 BM for the effective magnetic moment. The value was close to the spin-only moment of two unpaired electrons (2.84 BM). Moreover, the IR spectrum of L14 displayed broadband at 3545 cm-1 attributed to the group of phenolic OH, while this band disappeared from C14 spectrum, indicating the coordination vai the phenolic -OH. The involvement of the deprotonated phenolic moiety in complexes was also confirmed by the shift of v(C-O) stretching band observed at 1212 cm-1 in the free ligand to a lower frequency [80, 84]. The shift of v(C-O) band to a lower frequency suggested the weakening of v(C-O) and the formation of stronger M-O bond. The medium bands observed in frequencies 1641-1612 cm-1 in C17 were assigned to v(C=N) mode. The shift of v(C=N) vibration to C14 to a lower frequency suggested that the nitrogen atom of the ring contributed to the complexation as well. The lower v(C=N) frequency indicated stronger M-N bonding. The medium and low- intensity bands due to v(C-S) and v(C=N) modes of thiazole ring in the 1687-1690 and 741-744 cm-1 shifted to the lower frequency region (3136-3188 cm-1) which suggested the weakening of C-S and C=N bonds and formation of M-S bond. In the C14 complex, the bands observed at 270-294 and 320-328 cm-1 that were attributed to the v(M-N) and v(M-Cl) stretching vibrations, respectively [80, 84, 85]. The electronic spectrum of the C14, recorded in DMF, displayed two bands at 18.282 cm-1 (v1) and 22.272 cm-1 (v2) corresponding to an octahedral geometry. Regarding C15, the shift of azomethine v(-C=N-) group of the metal complex, to lower frequencies (1620-1576 cm-1) compared with the free ligand L15 (at 1629cm-1) could attribute to the coordination of two azomethine groups to metal ions. The weak bands in the two ranges (579-530 cm-1) and (459-439 cm-1) were assigned to the stretching frequencies of v(M-O) and v(M-N) bands, respectively, supporting that metal ion connected to the ligand via the phenolic oxygen and the azomethine nitrogen atoms of the ligand. The electronic spectrum of C15 showed an octahedral geometry. In the IR spectra of C16 and C17, the mode of chelation was supported by (i) the lower shift of (C=O), (ii) the disappearance of (C=O) with a simultaneous appearance of (C=N) at 1637 cm-1 and finally (iii) the lower shift of (C=N) to 1615 cm-1. Moreover, the magnetic momentum of C15 (μeff=2.54 BM) had a lower magnitude than the expected value for d3 system due to the spin-orbit coupling resulting in some mixing between the singlet ground state and triplet excited state.[39, 94, 95]. In C17, the magnetic momentum (μeff=5.34 BM) was higher than of the octahedral d3 system may be due to ferromagnetic interactions between metal ions. The coordination of Cr(III) with C18 determined via the carboxylate O atom. The latter was confirmed by the shift of the IR v(COO) stretching vibrations bands (symmetric and asymmetric) to 1589-1630 and 1404-1461 cm-1, respectively.[47.48]. The bands at 607 and 584 cm-1 found were attributed to COOH bending and NCOO bending of the carboxylate group [95]. The δ(ΝΗ2) in-plane and out of plane bending vibrations appeared at 1504-1511 and 769-789 cm-1 in metal complexes confirming the coordination of the amino group to the metal ions. The bands observed at 924-935 and 832-838 cm-1 were attributed to the coordination of water molecules [40]. The v(M-N) stretching vibrations appeared at 439-445 cm-1 for ternary metal complexes. In the C19 IR spectrum, the band of the carboxylic group was shifted to a lower frequency compared to that of L19, suggesting that the carboxylic group was coordinated with the metal ions. In addition, the stretching band of the primary amine group was also moved to a higher frequency indicating that the -NH2 was also involved in the coordination. The peaks were sharp, showing that the solid-state of the complex did not have intra / intermolecular hydrogen bonding, confirming that the carboxylate oxygen and the ligand's primary amino group were chelated to the metal ions. The UV-vis spectrum of L19, showed two bands at 239 and 318 nm, while when the ligand was complexed to the metal, a bathochromic shift was observed. The IR spectra of C20, showed small and medium bands at 1639-1652 cm-1 attributed to the v(C-O) stretching vibration of the aldehydic group. The bands found at the ranges 1604- 1610, 1428-1447, 1019-1025 and 599-681 cm-1 upon coordination suggesting the involvement of the lone pair on pyridyl nitrogen in bond formation with the metal ions. The appearance of the bands at 521-560 and 425-463 cm-1 was due to the coordination of metal ions with oxygen and nitrogen, respectively [42]. Regarding the C21 complex, a band at 1622 cm-1 in the L21 IR spectrum due to v(C=N) stretching vibration was shifted in lower frequencies (1604-1610 cm-1) after complexation. This shift was an obvious indication of the involvement of the azomethine nitrogen atoms in complex formation [93]. This was also supported by the appearance of the band at 451-470 cm-1, corresponding to the stretching vibration of the M-N bond. Bands at 569-559 cm-1 corresponded to M-O stretching vibrations. The characteristic frequencies of the coordinated nitrate group in C21, possessed three non-degenerate modes at 1472 cm-1 of v(NO2) asymmetry, 1352 cm-1 symmetry, and 855 cm-1 v(N-O) [28]. The v(C=O) stretching vibrations were observed at (1661-1668 cm-1) for L22 and C22, expressing the non- involvement of (C=O) amide group [96]. The IR spectra of the C22 demonstrated that the furan oxygen stretching vibrations were altered compared to those of ligand due to conformational changes. The furan oxygen stretching band was observed at 1226 cm-1 in the free Schiff base L22, which was shifted to higher wavenumbers in C22, indicating the participation of the furan oxygen in coordination [30, 33, 34]. New bands appeared in the spectra of C22 in the regions of 467-470 and 420-432 cm-1, assigned to v(M-O) and v(M-N) stretching vibrations, respectively [97]. The electronic spectrum of C22 gave three bands at 23.866, 19.685 and 17.543 cm-1, indicating an octahedral structure [98, 44]. The IR spectrum of C23 presented a shift of the N-C vibrating band compared to L23. These results indicated a greater rigidity in the chemical bonds in C26, confirming the metal’s coordination with the azole's north. Bands were also observed at 3435 and 3429 cm-1 that corresponded to the stretches of the bond (O-H) in water molecules. For L23, a band between 200 and 300 nm corresponded to the transitions between the π-π* orbitals was observed in the UV-vis spectrum. Besides, for C23, the most intense band was not shifted relative to the band of L23. This peak was increased in intensity and this was attributed to the interaction of the orbitals of the ligand with those of the metal [98, 99]. Moreover, a medium intensity band of v(C=N) (1630-1608 cm-1) was observed for C24, which was shifted to lower frequencies compared to that of L24, indicating the coordination via the nitrogen. This Cr (II) complex showed broadband between 3592 and 3432 cm-1, which indicated the presence of lattice or coordinated water in this complex. The presence of a medium sharp band in this complex of Cr (II) in the region of 762-712 cm-1 indicated the coordination of water molecules in C24. The disappearance of the characteristic band of v(COOH) of the amino acid at 1722 cm-1 which was present in L24 and the appearance of two new absorption bands around 1592-1526 and 1398-1342 cm-1, corresponding to v(COO-) asymmetric and symmetric, supporting the participation of the carboxylate oxygen of this group in the chelation. The observed negative shift of v(N-H) of glycyl glycine from 3320 to 3208 cm-1 of C24, in comparison to L24, confirmed the coordination of amide nitrogen to Cr (II). Besides this, the disappearance of v(C=O) of the β-diketone and the appearance of a new band at 1568 cm-1 assignable to v(C-O) suggested that the carbonyl group involved in the coordination in the enol form through deprotonation. Further, the metal-ligand bonds through the carboxyl oxygen and the imino nitrogen of L24 were confirmed by the far-IR spectra. The latter spectrum showed non-ligand bands corresponding to v(Cr-O) vibrations (at 470-450) and v(Cr-N) vibrations (at 544-508 cm-1) [47, 59]. Moreover, the phenolic v(C-O) stretching vibrations presented at 1156 cm-1 in L25 was shifted to lower wavenumbers in C25, indicating coordination of the phenolic oxygen to the metal ion. In addition, the IR spectrum of L25 exhibited strong bands at 1604 and 1570 cm-1 assignable to v(C=N). In the spectrum of the corresponding C25, these bands shifted to a lower frequency (by about 15-20 cm-1), suggesting the coordination of the azomethine nitrogen to the metal centre. The absorption band of v(C=S) in the spectrum of L25 was absent since the sulfur atom was alkylated with iodomethane, leading to the appearance of a new absorption band at 682 cm-1 correspondings to delv(C-S) [79]. This band was not shifted, indicating that the sulfur atom was not involved in coordination to the metal ion [17]. For C26, the appearance of absorption bands in the region of 3500-3200 cm-1 was indicative of the presence of water molecules in its structure. The IR spectrum of L26 displayed a strong band at 1617 cm-1 due to the azomethine group v(CH=N) stretching frequency. The band was shifted to a higher frequency for C26 at 1615-1651 cm-1 indicating coordination through the azomethine-nitrogen with the metal ion. The spectrum of L26 also showed strong bands at 1276 and 1386 cm-1 assigned to v(C-O) phenolic and v(C-N) stretching vibrations of the ligand, respectively [73, 74]. However, after complexation via oxygen and nitrogen to the metal ion, these bands were observed at the regions of 1220-1284 cm-1 and 1365-1383 cm-1 for v(C-O) and v(C-N), respectively. The bands observed for C26, at 537-590 cm-1 and 450-4474 cm-1 were metal sensitive and were assigned to v(M-O) and v(M-N), respectively [108]. The v(M-O) stretching vibrations of coordinated water were found at 520-574 cm-1. The magnetic moment of the octahedral C26 molecule was found 3.58 Β.Μ., a value agreed with the presence of three unpaired electrons. The representative IR spectra vibrations for C27 were at 1667 cm-1 for v(C=O), 839.6 cm-1 for v(C-N) aromatic and 581.7 cm-1 for v(Cr-O). For C28, bands at 1660.8 cm-1 for v(C=O), 1476.6 cm-1 for v(C-C) aromatic stretching vibrations and 3090.5 cm-1 for v(Cr-O) were found. Furthermore, characteristic IR spectrum bands for C29 showed at 1669.8 cm-1 v(C=O), 1602.7 cm-1 v(C-C) aromatic and 3512.9 cm-1 v(Cr- O). For C30, three bands (1709.2, 1681.5 and 1615.5 cm-1), corresponded to C=O asymmetrical stretching vibrations were observed. Another peak at 3000 cm-1 indicated the H2O in the coordination sphere, while the peak at 550.0 cm-1 corresponded to the coordinated oxygen with the metal ion. The asymmetrical stretching vibrations of the C=O bond for C31 were found at 1709.8 cm-1, 1690.6 and 1659.5 cm-1. The stretching vibrations of v(C-O) and v(C-C) were found at 1390.0 cm-1, while the peak at 3000 cm-1 indicated the H2O in the coordination sphere. Moreover, the band at 550.0 cm-1 corresponded to the coordinated oxygen with the metal ion [51]. Finally, the IR spectrum of L32 displayed fundamental spectral bands located at 3397, 3290, 1623, 1312, 1074, and 491 cm-1 characterized to v(NH2), symmetric and asymmetric, and v(C-NH2). Also, the different vibration modes of v(C=N), v(N-N) of pyrazole ring were confirmed by bands at 3190, 3114, 1458, 1607 and 1127 cm-1 [27]. The stretching deformation and bending vibrations modes of C-H of methyl groups were associated with the bands appeared at 2971, 2890, 1407 and 826 cm-1. The spectrum of L32 gave bands at 3040, 1520, 1023 and 615,579 cm-1 referred to stretching and bending of C-H in-plane of the pyridine ring [29]. The infrared spectra of C32 showed the v(NH2) symmetric and asymmetric, undergo shift towards higher wavenumber by 14-59 and 19-21 cm-1, respectively relative to L32. The spectra of C32 gave a new band at (absent in L35) in the range of 526-558 cm-1 assignable to v(M-N) [92, 32]. In addition, the appearance of two bands at 1489 and 1288 cm-1 assignable to v(NO -) symmetric and asymmetric, indicated that the NO - group coordinated as a monodentate moiety [43]. The pyrazole ring v(C= N), v(N-N) and pyridine ring bending in and out of an airplane were associated with a very slight shift in their position. This omitted the non-involvement of the imine, NH, and pyridine ring nitrogen atoms in coordination with the metal ion. The observed (NH) group change and some pyridine ring bands attributed to the presence of inter / intramolecular hydrogen bonding after complexation. This argument showed that the ligand functioned as a monodentate neutral and interacted with metal ions through the amino group's nitrogen atom. The electronic spectrum of L32 gave four bands at 330, 360, 415 and 463 nm assigned to π-π* and n-π* transitions. The band observed at 463 nm in the spectrum of L32, assigned to n-π* transition, suffered a redshift to 465-480 nm due to a combination of LMCT with d-d transitions [53].
4. ANTIBACTERIAL STUDIES
This section will describe the antibacterial activity of all complexes presented before. Reddy et al. [25] studied the antibacterial activities of macrocyclic Cr (III) complexes (C1, C2, C3) as well as their ligands and compared their activity with that of known antibacterial drugs, streptomycin, ampicillin and rifampicin. Preliminary screening was performed at fixed concentrations of 1000 μg / ml for complex molecules C1, C2 and C3. The molecules C1, C2 and C3 showed greater activity than the corresponding molecules L1, L2, and L3. Comparing their activity with that of known antibacterial agents, the activity of these three molecules against gram-positive bacteria (Bacillus subtilis, Staphylococcus aureus) and gram-negative (Escherichia coli, Klebsiella pneumonia) was found to be more successful. The antimicrobial activity of L1, L2 and L3, as well as their corresponding complexes C1, C2 and C3, was attributed to the presence of the C=N group (Schiff base). It has also been proposed that chelation decreased the polarity of the metal ion mainly due to partial sharing of its positive charge with a donor group throughout the entire chelate ring system. This chelation process, therefore, enhanced the lipophilic nature of the central metal ion, which in turn facilitated its permeation through the lipid layer of the membrane, thereby allowing the metal complex to more easily cross the bacterial membrane, increasing the complexes' activity. In addition, many other factors, such as solubility, dipole moment, conductivity that influenced by a metal ion, could contribute to the antibacterial activity of C1, C2, and C3. In addition, the involvement of a thio group contributed to the antibacterial activity as well [25]. The C4 was evaluated against two gram-positive, two gram-negative bacteria and four fungi (two mold and two yeast strains). Standard antibiotics, namely Ciproflaxin and standard antifungal drug Fluconazole were used for comparison. C4 presented strong antibacterial activity against gram-positive bacteria (S. aureus, B. subtilis) and strong antifungal activity toward the yeast S. cerevisiae and molds A. niger and A. fumigatus. However, C4 did not work against gram-negative bacteria (E. coli and P. aeruginosa) and yeast C. Albicans [26] L5 and C5 were evaluated on S. Aurores and E. Coli where C5 was found more effective compared to L5. The increased antimicrobial activity of the metal chelates can be explained based on Overtone’s concept and Tweedy’s chelation theory [71]. On chelation, the delocalisation of pi-electrons over the whole chelate ring increases the liposolubility of the complexes. The hydrocarbon tail serves as a lipophilic group to move the compound through the cell's semi-permeable membrane and block metal-binding sites within microorganism enzymes. The Schiff base ligand L6 and it chelate C6, were tested for their inhibitory effects on the growth of bacteria: S. aureus and E. coli and fungi: A. flavus and A. niger fungi since such organisms can achieve resistance to antibiotics through biochemical and morphological modification [45, 46]. The antimicrobial activity was tested by the agar well diffusion method. The antibacterial results indicated that C6 was more active against the same microorganisms than the parent L6. In addition, C6 showed poor behaviour against bacterial species compared to standard one's Streptomycin, while it exhibited better activity against the fungi species when compared with Chlorometazole. This was attribute to the fact that the chelation makes the complex's ability to cross a cell membrane easier. The variability in the effectiveness of various compounds against different organisms depends on the permeability of microbial cells or variations in microbial cell ribosomes [72]. Regarding the C7 molecule, it showed more inhibitory effects [90] than the parent L7 molecule. The latter was attributed to the fact that complexes may deactivate different cellular enzymes, which play a vital role in the microorganism metabolic pathways. It was also suggested that the toxicant's ultimate action was the denaturation of one or more cell proteins, impairing normal cellular processes as a consequence. Other factors that also increased the antimicrobial activity were the solubility, conductivity and length of bond between metal and ligand. C7 exhibited better antibacterial activity toward E. coli, rather than S. aureus, Proteus Vulgaris, Pseudomonas and Klebsiella [11]. The activity of L8 and C8 was tested against some human pathogenic bacteria, including gram-positive (S. aureus and B. cereus) and gram-negative (E. coli, P. aeruginosa, K. pneumoniae, and P. Vulgaris). In particular, C8 was more active against bacteria and specifically, C8 indicated similar antibacterial activity with the antibacterial drug Ciproflaxin, for P. Vulgaris, P. aeruginosa and S. aureus bacteria [31]. The gram-positive and gram-negative bacteria utilized for the antibacterial study of C9 consisted of S. aureus, B. thuringiensis, P. aeruginosa and E. coli. C9, showed good bacterial inhibition against gram-negative bacteria, in comparison with Centamycin as a reference drug [24]. The antimicrobial activity of L10 and C10 was tested in vitro against multi-resistant bacterial strains isolated under clinical conditions. The bacteria strains used, namely: E. coli, P. aeruginosa and S. aureus are resistant to oxacillin, cefoxitin, ampicillin, erythromycin, clindamycin, kanamycin, tetracycline, tobramycin, septrin, and chloramphenicol. L10 showed negligible activity against E. coli and S. aureus, but moderate activity against P. aeruginosa. On the other hand, C10 showed improved activity against E. coli and P. aeruginosa [18]. The hydrazine-based L11 and its metal complex (C11) were screened for their antimicrobial activities. It is observed that C11 had higher activity than the free ligand L11. They were tested against gram-positive (Staphylococcus aureus, Streptococcus pyogenic) bacteria, gram-negative bacteria (Pseudomonas phaseolicola, Pseudomonas fluorescens) and fungi (Fusarium oxysporum, Aspergillus fumigatus). C11 presented mild activity against gram-positive and gram-negative bacteria, while it was showed low activity against fungi [36]. In Mueller Hinton-Agar media, the in vitro antimicrobial activity of L12, L13 with their complexes, C12, C13, was tested against Staphylococcus aureus (gram-positive) and Escherichia coli (gram-negative). To assess the antibacterial activity of the synthesized compounds, the traditional method of surface diffusion was followed. The antibacterial activity of L12 and L13 was found moderate in comparison with C12 and C13 [35]. L14 and C14 were tested against Staphylococcus aureus, Streptococcus pyogenes (Gram-positive), Pseudomonas phaseolicola, Escherichia coli, Pseudomonas purida (Gram-negative), Fusarium oxysporum, Aspergillus fumigatus and Candida albicans (Fungi). L14 wasn’t active at all against Pseudomonas phaseolicola and Escherichia coli, whereas on the rest of bacteria and fungi showed only moderate activity. Chloramphenicol, Cephalothin and Cycloheximide were used as a standard reference for Gram-positive bacteria, Gram-negative bacteria, and fungi, respectively. C14 showed excellent activity (100% inhibition) on Gram-positive bacteria, and good activity (60-75% inhibition) to the rest of Gram-negative and fungi, except Aspergillus, fumigatus, where the activity was significant (30-50%) [37]. The diffusion agar technique was also used to evaluate the antimicrobial activity of C15 as well. C15 showed very high inhibition levels on S. aureus, S. pneumoniae, B. subtilis (Gram-positive), P. aeruginosa, K. pneumoniae, E. coli (Gram-negative), A. fumigatus and C. Albicans (Fungi) [38]. L16, L17 and their complexes C16 and C17, were screened separately for their antimicrobial activity against E. coli and B. subtilis. The activity of L16, L17, C16 and C17 was compared with the activity of common standard antibiotic Ampicillin. All ligands and complexes presented mild activity against E. coli bacteria and no action against B. Subtile. The negative results were attributed either to the failure of the complexes to penetrate into the cell membranes of the gram-positive bacteria, or their inactivation through the unknown cellular mechanism, i.e., bacterial enzymes [39]. C18 was generally more active than the L18. C18 was more susceptible to the gram-negative bacteria due to their thin peptidoglycan layer making it more permeable to the complexes. This enhancement in antibacterial activity was rationalized based on the partial sharing of the positive charge of metal ions with donor groups [86-90]. This may support the argument that inhibition of biological synthesis is caused by some type of biomolecular binding to metal ions, or interchelation or electrostatic interactions. Therefore some complexes have a higher antimicrobial activity than the free ligand [40]. Compounds L19 and C19 were tested for their antimicrobial activity against Bacillus species, BSER and BLF1, with C19 showing better activity against BSER. C19 also demonstrated fungicidal activity towards F. oxysporum and M. phaseolina [41]. The azo-free ligand L20, and its metal complex (C20) were screened for antimicrobial activity against two strains of Gram-positive bacteria, B. subtilis and S. aureus, and two strains of Gram-negative bacteria, E. coli and N. gonorrhoeae, as well as C. albicans fungus. The data were compared with standard antibiotic Amikacin for antibacterial activity and Ketoconazole for antifungal activity. The in vitro antibacterial activities showed that C20 had higher antimicrobial activity in comparison with that of the azo L20. Regarding the antifungal activity, L20 had no activity against the fungus C. albicans, while C20 presented significant activity better than that presented by the standard antifungal drug. The latter leads the research toward such azo ligand as principle cytotoxic species [42, 56]. The in vitro antimicrobial activity of the L21 and its complex against three selected bacteria Escherichia coli, Bacillus subtilis and Staphylococcus aureus as well as three fungi Aspergillus flavus, Candida albicans and Trichophyton rubrum was evaluated. L21 and C21 displayed good biological activity with the micro-organism, while C21 was more efficient than L21 [28] L22 and C22 were tested against Aspergillus fumigatus and Candida albicans (fungi), Streptococcus pneumonia and Bacillus subtilis (gram-positive bacteria) and Pseudomonas aeruginosa and Escherichia coli (gram-negative bacteria). The activity of L22 and C22 was compared to Amphotericin, Ampicillin and Gentamicin. In all cases, C22 was more active than L22, while its activity was similar to those of the antibiotic drugs [44]. The antibacterial activity of L23 and C23 were studied against Staphylococcus aureus, Salmonella Typhimurium and Escherichia coli. Standard drugs Ampicillin, Gentamicin, Amphotericin B and Itraconazole, were used for comparison. Bacterial species were more resistant to treatments with C23. In addition to this, L23 was more effective against fungi [102] L24 and C24 were tested against Bacillus subtilis, Staphylococcus aureus, Escherichia coli and Klebsiella pneumonia showing high antimicrobial activity, while C24 was more active than L24. It was concluded that the ligand with N and O donor systems might have inhibited enzyme production. The antibacterial and anti-fungal properties of L24 and C24 were tested against S. aureus and E.faecalis (gram-positive), E. coli and S. abony (gram-negative) bacteria and C. Albicans (fungi). The study showed that L24 and C24 compounds had bacteriostatic and bactericidal activities. In particular, C24 was more active than the L24. The activity of C24 was less pronounced towards gram-negative microorganisms than towards gram-positive bacteria and fungi. Nevertheless, the antibacterial and antifungal efficiency of the C24 molecule was lower than those presented by furacillinum and nystatin that are used in medical practice [47]. The biological activity of L26 and C26 was tested in vitro against two fungi species, Aspergillus fumigates and Candida albicans, two gram-positive bacteria, Staphylococcus aureus and Bacillus subtilis and three gram-negative bacteria, Salmonella SP., Escherichia coli, and Pseudomonas aeuginosa. Ampicillin and Gentamycin were used as reference compounds for antibacterial activity, while Amphotericin B was used as a reference compound for antifungal activity. The increased activity of metal chelates was explained based on the chelation theory described previously [60-63]. The activity of C26 was similar to the reference compounds for gram-negative bacteria. The compounds C27 to C31 [51] were assessed against six bacteria species namely: B. subtilis, E. coli, E. faecalis, P. aeruginosa, S. aureus and S. epidermidis. Compounds C28, C30 and C31 showed neither bacteriostatic nor bactericidal activity. The compounds C27 and C29 exhibited bacteriostatic activity against only some bacteria. C27 was active against Bacillus subtilis, Escherichia coli and Enterococcus faecalis, while C29 was efficient against Bacillus subtilis, Escherichia coli and Pseudomonas aeruginosa. However, none of these two compounds exhibited any bactericidal activity. Finally, using the paper disk diffusion method, L32 and its metal complex (C32) were evaluated for their antimicrobial activity. These compounds were screened against Staphylococcus aureus and Bacillus cereus as gram-positive bacteria and Escherichia coli and Pseudomonas aeruginosa as gram-negative bacteria. Both presented low activity towards gram-positive bacteria and no activity towards gram-positive bacteria. The microorganisms tested for each complex presented are listed in Table 1 [53].
| Complex Molecule | Microorganism Tested | Reference |
|---|---|---|
| C1 | B. subtilis, S. aureus E. coli, K. pneumonia | [25] |
| C2 | ||
| C3 | ||
| C4 | S. aureus, B. subtilis, S. cerevisiae, A. niger, A. fumigatus, E. coli, P. aeruginosa, C. Albicans | [26] |
| C5 | S. Aurores, E. Coli | [19] |
| C6 | S. Aureus, E. coli,A. flavus, A. niger | [21] |
| C7 | E. coli, S. aureus, P. Vulgaris, Pseudomonas, Klebsiella | [11] |
| C8 | S. Aureus, B. cereus, E. coli, P. aeruginosa, K. pneumoniae, P. Vulgaris | [31] |
| C9 | P. Vulgaris, P. aeruginosa, S. aureus | [24] |
| C10 | E. coli, P. aeruginosa, S. aureus | [18] |
| C11 | S. aureus, S. pyogenic, P. phaseolicola, P. fluorescens, F. | [36] |
| oxysporum, A. fumigatus | ||
| C12 | S. aureus, E. coli | [35] |
| C13 | ||
| C14 | S. aureus, S. pyogenes, P. phaseolicola, E. coli, P. purida, F. oxysporum, A. fumigatus, C. albicans | [37] |
| C15 |
S. aureus, S. pneumoniae, B. subtilis, P. aeruginosa, K. pneumoniae, E. coli, A. fumigatus, C. Albicans |
[38] |
| C16 | E. Coli, B. subtilis | [39] |
| C17 | ||
| C18 | B. subtilis, E. coli, N. gonorrhoeae, S. aureus, C. albicans | [40] |
| C19 | Bacillus species, F. oxysporum, M. phaseolina | [41] |
| C20 | B. Subtilis, S. aureus, E. coli, N. gonorrhoeae, C. albicans | [42] |
| C21 | E. coli, B. subtilis, S. aureus, A. flavus, C. albicans, T. rubrum | [28] |
| C22 |
A. fumigatus, C. albicans, S. pneumoniae, B. subtilis, P. aeruginosa, E. coli |
[44] |
| C23 | S. aureus, S. Typhimurium, E. coli | [102] |
| C24 | B. subtilis, S. aureus, E. coli, K. pneumonia | [47] |
| C25 | S. aureus, E.faecalis, E. coli, S. abony, C. Albicans | [17] |
| C26 | A. Fumigates, C. albicans, S. aureus and B. subtilis, Salmonella SP., E coli, P. aeuginosa | [50] |
| C27 | B. subtilis, E. coli, E. faecalis, P. aeruginosa, S. aureus, and S. epidermidis. | [51] |
| C28 | ||
| C29 | ||
| C30 | ||
| C31 | ||
| C32 | S. Aureus, B. cereus, E. coli, P. aeruginosa | [53] |
CONCLUSION
Nowadays, one of the greatest challenges that scientists are facing is the resistance that several bacteria developed over the established antibiotics. Novel compounds should be developed and evaluated against different bacterial. Metal-based drugs are among the most promising compounds that can be used in that direction.
This review presents the synthesis and structural characteristics of both 32 molecules (free ligands) and their corresponding Cr(III)-complexes, as well as their antimicrobial activity against several species of bacteria. Based on this review, metal coordination to several ligands improved the antimicrobial activity of metals or ligands dramatically. Cr(III)-complexes seem to present a very promising antimicrobial activity against several bacteria. Sometimes the antimicrobial activity of Cr(III)-complexes was comparable or better than that of clinical drugs. The mechanism of action of these complexes has not been completely understood yet. Proposed pathways include the induction of oxidative stress on bacteria, which leads to the disruption of their cell membrane. Moreover, the improved efficiency was attributed to the increase of permeability of the cell membrane of the microbes via the introduction of ligands with hydrophilic moieties or other groups, while more challenge explanation relies on the promotion of protein dysfunction, the production of ROS, the genotoxicity and the interference with nutrient assimilation.
Despite these promising results, Cr(III)-complexes have not systematically studied compared to other transition metal-complexes. Many challenges are present regarding the synthesis of new Cr-complexes with exceptional antimicrobial activity that could replace the existing drugs eliminating the problem of microbial resistance.
CONSENT FOR PUBLICATION
All patients participated on a voluntary basis and gave their informed consent.
FUNDING
None.
CONFLICT OF INTEREST
The author declares no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
Declared none.

