All published articles of this journal are available on ScienceDirect.

HPLC and HPLC-MS Analysis of Intestinal Elimination and Phase 2 Metabolism of 4'-hydroxy-4-Methoxychalcone and its Bis-Mannich Analog In The Rat
Authors Info & Affiliations
Abstract
Aims:
The aim was to study the elimination and Phase 2 biotransformation of 4'-hydroxy-4-methoxychalcone (1) and its bis-Mannich analog (2) in the small intestine of the rat.
Background:
Earlier studies indicated that chalcones are promising starting points for drug design. Aminomethylation of drugs is considered to improve their delivery into the human body.
Objectives:
To set up validated HPLC-UV methods to quantitate the investigated chalcones in the rat intestinal perfusates. Comparison of intestinal disappearance and Phase 2 metabolic profile of the 4’-hydroxychalcone (1) and a bis-Mannich analog (2).
Methods:
Chalcones 1 and 2 were luminally perfused in the small intestine of rats at a concentration of 240 μM and 280 μM, respectively. Analysis of the collected intestinal perfusate samples was performed by a validated HPLC-UV method. Using HPLC-MS, the samples were analyzed for Phase 2 metabolites as well.
Results:
Elimination kinetics of the two 4’-hydroxychalcones displayed characteristic differences having the nonpolar chalcone 1 higher elimination rate over the 90-minute ex vivo experiments. HPLC-MS analysis of the perfusates indicated the presence of glucuronide, sulfate, and glutathione conjugates in the parent molecules. Intestinal disappearance and sulfation of the bis-Mannich derivative 2 showed characteristic differences compared to 1
Conclusion:
The results demonstrate, to the best of our knowledge, for the first time, how the title structural modification of phenolic chalcones affects intestinal elimination and Phase 2 metabolism of the compounds
Highlights:
Study on ex vivo intestinal elimination of a 4'-hydroxy-4-methoxychalcone and its bis-Mannich analog.
Development of validated HPLC-UV methods for quantitation of 4’-hydroxychalcone derivatives in rat intestinal perfusates.
HPLC-MS identification of Phase 2 metabolites of 4’-hydroxychalcones in rat intestinal perfusates.
1. INTRODUCTION
Hydroxychalcones are intermediary compounds of the biosynthetic pathway of natural flavonoids [1]. The wide range of biological activities of both naturally occurring and synthetic analogs, among others, are cytotoxic [2, 3], antitumor [4], anti-inflammatory [5], and chemopreventive (antioxidant) [6, 7] properties are well documented in the literature [8-11]. However, only a few absorption and metabolic studies can support the relevance of chalcones as drug candidates [12-15].
Aminoalkylation reactions can change the lipophilicity of a drug leading to improved drug delivery to the active sites in the human body [16]. Solubility of the Mannich bases in water could be increased due to their protonation to form the respective ammonium salts [17]. They can act as prodrugs forming reactive derivatives via deaminomethylation or deamination [18]. Furthermore, transport systems facilitate the absorption of tertiary amines from the small intestine [19, 20]. Previously, ortho aminoalkylation of the phenolic ring in chalcones resulted in improved pharmacodynamic properties [21-24].
Recent literature has shown phenolic Mannich bases derived from chalcone analogs to have remarkable cytotoxic potencies toward cancer cell lines [25-29]. Earlier, we reported on the synthesis, stereochemistry, and GSH-reactivity of 4’-hydroxychalcones and their bis-Mannich base derivatives [30]. It was found that the Mannich-base derivatives showed higher GSH reactivity than two respective 4’-hydroxychalcones. Previous investigations showed chalcone 1 as a potent inhibitor of MDA-MB-231 cells (IC50 = 3.8 µM) [31]. Furthermore, the bis-Mannich hydroxychalcone 2 is a relatively potent inhibitor (IC50 = 17.9 μM) of PC-3 cells [18]. It is also important to emphasize that the bioactivity of compound 2 also extended to its higher in vitro reactivity with the major thiol intracellular, the glutathione, compared to its hydroxychalcone precursor in a recent study performed by our research group [30]. Furthermore, compound 2 proved to induce a weak cleavage of plasmid DNA [32].
Karakaya summarized the factors that affect the absorption, bioavailability, and metabolism of food phenolic compounds [33]. It was found that partition coefficients seemed to play an influential role in the absorption of phenolics regardless of whether they have sugar or organic acid substitution in their structures. Hydrophobic phenolics having sugar (other than rhamnose) substitution in their structure are absorbed through the small intestine by the cytosolic beta-glucosidase/galactosidase. Their absorption is also related to the specificity of carriers [33]. In addition, oral drug absorption was similar in rats and humans, making permeability studies in rat intestines helpful in predicting in vivo absorption [34, 35].
The present investigations were designed to study the intestinal elimination and metabolism of the 4’-hydroxy-4-methoxychalcone 1 and its bis-Mannich analog 2 in rat proximal jejunum, which was perfused with isotonic medium containing 240 μM (0.061 mg/mL) of 1 or 280 μM (0.136 mg/mL) of 2. This type of drug perfusion basically corresponds to oral administration [35]. The perfused rat intestinal model is widely used for studying the absorption of drugs and, more recently, natural products such as flavonoids [36].
Using this method, we aimed to compare luminal elimination kinetics and metabolism of 1 and its bis-Mannich analog 2 (Fig. 1) to see how introducing a non-hydrolyzable polar moiety affects absorption and metabolism of the chalcone structure. Validated analytical methods have been developed for quantitative comparison to determine the two parent compounds in the perfusates. Metabolites were planned to be identified by HPLC-MS.
2. MATERIALS AND METHODS
2.1. Chemicals
Sodium chloride, sodium dihydrogen phosphate, disodium hydrogen phosphate dihydrate, 2,5-dihydroxybenzoic acid (2,5-DHB), polyethylene glycol (PEG) 400, formic acid ≥ 98%, and perchloric acid 70% were obtained from Sigma-Aldrich (Budapest, Hungary). HPLC-grade methanol was obtained from VWR International Kft. (Debrecen, Hungary), potassium hydroxide pellets were purchased from Merck Kraal (Darmstadt, Germany). Potassium chloride, magnesium sulfate, calcium chloride dihydrate, glucose, mannitol, and tris(hydroxymethyl)aminomethane (TRIS) were obtained from VWR International Kft. (Debrecen, Hungary). Distilled water was purified in the Institute of Pharmaceutical Chemistry, University of Pécs, using a Millipore Direct-QTM system. The standard isotonic perfusion medium had the following composition (mmol/L): NaCl 96.4, KCl 7.0, CaCl2 3.0, MgSO4 1.0, sodium phosphate buffer (pH 7.4) 0.9, TRIS buffer (pH 7.4) 29.5, glucose 14.0, mannitol 14.0.
2.2. Synthesis of Compounds 1 and 2
The 4'-hydroxy-4-methoxychalcone (1) [37] and the bis-Mannich chalcone (2) [30] were synthesized as published earlier. Details of the synthesis and the analytical data of the compounds are summarized in the Supplementary materials.
2.3. Perfusion Experiments and Sample Preparation
The experiments were performed according to the protocol used before [34, 38, 39]. Male Wistar rats (weighing 220-280 g) were used. The animals fasted for 18–20 h before the experiments; water was provided ad libitum. The animals were anesthetized with urethane (1.2 g/kg, i.p.). The abdomen was opened by a midline incision, and a jejunal loop (length about 10 cm) was isolated and cannulated. Body temperature was maintained at 37°C. The lumen of the jejunal loop was gently flushed with a warmed isotonic solution to remove digesta and food residues and then blown empty with 4–5 mL of air. The intestinal perfusion was carried out by a peristaltic pump (flow rate: 13 mL/min) with 16.5 mL of 240 μM (0.061 mg/mL) chalcone 1 or 280 μM (0.136 mg/mL) chalcone 2 dissolved in the isotonic perfusion medium (containing 3.5 v/v % PEG-400 in the case of 1), in a recirculation mode for 90 minutes. During the 90 min period of the perfusion, 250 μL perfusion
samples were collected at the 0, 0.5, 1, 3, 5, 10, 15, 22, 29, 36, 45, 60, 75, and 90 minute time points. The samples were stored at -20 °C until analysis. Experiments were performed using three animals with both compounds.
Before analysis, the temperature of the samples (250 μL) is allowed to rise to ambient temperature. Then 10 μL of perchloric acid solution (3.88 M). and 10 µL of 1.4 mg/mL (9.1 mM) solution of 2,5-DHB in methanol (as internal standard) were added to each sample. The samples were vortex mixed for 40 s and centrifuged at 10000 × g for 10 min. The supernatant was collected, and 5 µL of potassium hydroxide solution (3.96 M) was added (pH 3). After vortex mixing for 40 s, the samples were centrifuged at 10000 × g for 2 min and analyzed. (Final concentration of 2,5-DHB is 50 μg/mL.)
2.4. Preparation of Standard Solutions
A stock solution of 1 was prepared by dissolving 6.1 mg of chalcone 1 in 5.0 mL of a mixture of PEG-400 and Krebs-Tris buffer solution (7:1, v/v), while a stock solution of 2 was prepared by dissolving 6.25 mg substance in 5.0 mL of distilled water.
For the animal experiments with chalcone 1, the stock solution was diluted with an isotonic perfusion medium to obtain a 0.061 mg/mL (3.5 v/v % PEG-400) solution; and the stock solution of 2 was diluted with the perfusion medium to get a 0.136 mg/mL solution.
Working standard solutions for the validation studies were prepared by diluting the appropriate volume of stock solutions to 250 μL by blank perfusate (perfusate obtained from perfusion of the isotonic perfusion medium free of the drugs) and treated them as the samples.
2.5. HPLC-UV Analysis
HPLC analyses were performed with an Agilent 1100 (Agilent Technologies, Waldbronn, Germany) integrated HPLC system equipped with a quaternary pump, a degasser, an autosampler, and a variable wavelength UV–Vis detector. Compounds were separated on a Thermo ScientificTM AccucoreTM RP-MS (150 mm x 2.1 mm, particle size 2.6 µm) column equipped with a guard cartridge. Data were recorded and evaluated using Agilent ChemStation (Rev.A.10.02) software.
The mobile phase consisted of water-0.1% formic acid (A) and methanol-0.1% formic acid (B). The mobile phases were degassed in an ultrasonic bath (Realsonic cleaner) and filtered through a ROBU-GLASS filter (Por.4) before use. The temperature was set at 45 °C, the flow rate was 0.25 mL/min, and the injection volume was 5 µL.
For samples of chalcone 1 (UV detection at 350 nm), the following gradient profile was used: 80% to 75% of A up to 5 min, 35% of A up to 25 min, 80% of A up to 27 min, 80% of A up to 50 min (Method 1). For samples of chalcone 2 (UV detection at 330 nm), the following gradient profile was used: 90% A for 6 min, 37% of A up to 8 min, 37% of A up to 11 min, 90% of A up to 11.5 min, 90% of A up to 29 min (Method 2).
2.6. LC-MS Analysis
2.6.1. Sample Preparation
For HPLC-MS analysis of chalcone 1 samples, 0.2 mL of perfusate was mixed with 50 μL of 30% ammonia solution and vortexed. Then, solid-phase extraction was performed using an Oasis MAX 3cc Vac cartridge (mixed-mode anion exchange reversed-phase sorbent for acids, 60 mg sorbent per cartridge), previously conditioned with 1 mL of methanol and 1 mL of water. After the sample was loaded onto the cartridge, the cartridge was washed with 1 mL of 0.1% formic acid solution-methanol (50/50, v/v), and the sorbent bed was dried by applying vacuum for 10 seconds. Elution was achieved using 2 mL of 2% formic acid in methanol. The eluate was evaporated to dryness using nitrogen gas, and the residue was reconstituted in 50 μL of a mixture (80%/20%, v/v) of formic acid in water (0.1%, v/v) and formic acid in methanol (0.1%, v/v).
For LC-MS analysis of sample 2, 0.2 mL of perfusate was mixed with 50 μL of 2% formic acid solution and vortex mixed. Then, solid-phase extraction was done using Isolute HCX mixed-mode SPE sorbent (nonpolar C8 with cation exchange, 50 mg/1 mL), previously conditioned with 1 mL of methanol and 1 mL of water. After the sample was loaded onto the cartridge, the cartridge was washed with 1 mL of a mixture (50/50, v/v) of 0.1% formic acid in methanol; and the sorbent bed was dried applying vacuum for 30 seconds. Elution was achieved using 2 mL of the mixture of 25% ammonia solution and methanol (5/95, v/v). The eluate was evaporated to dryness using nitrogen gas, and the residue was reconstituted in 50 μL of a mixture (90%/10%, v/v) of formic acid in water (0.1%, v/v) and formic acid in methanol (0.1%, v/v).
In the lack of the respective glucuronic acid-. Sulfate-, and GSH-conjugates, determination of recovery data, and HPLC analyses with validated HPLC methods could not be performed. Accordingly, the samples were used exclusively for the MS detection and identification of the expected Phase 2 metabolites.
2.6.2. HPLC-MS Conditions
The integrated high-performance liquid chromatography system (Jasco) - qualified and verified according to the pharmaceutical requirements - was equipped with an HPLC pump (PU-980), a degasser, a manual injector (RHEODYNE 7725i) with a 5 μL loop, a column oven, and a diode-array detector (MD-2010). Data were recorded and evaluated by ChromNav software (ver. No. 1.21). The Jasco HPLC-system was connected to a Waters Xevo TQ-S Mass Detector. MS data were recorded and assessed by MassLynx V4.1 (Waters) software. Compounds were separated on a Thermo ScientificTM AccucoreTM RP-MS (150 mm x 2.1 mm, particle size 2.6 µm) analytical column
The HPLC mobile phase consisted of methanol:water-formic acid (15:85-0.05%) (A) and methanol-water-formic acid (90:10-0.05%) (B). The mobile phases were degassed in an ultrasonic bath (Realsonic cleaner) and filtered through a ROBU-GLASS filter (Por.4) before use. The temperature was set at 45 °C, the flow rate was 0.25 mL/min, and the injection volume was 5 µL. Chromatography was performed using the gradient Method 1 and Method 2 as described in section 2.5.
The MS measurements were performed both in positive and negative ion mode. The ESI source was operated with a spray voltage of 3.00 kV; the cone voltage was 30 V. Desolvation gas was delivered at 500 L/hour and thermostatted at 500 °C. Cone gas was delivered at 150 L/hour. Collision gas pressure was 0.13 bar. Full-range mass spectra (80-1000 m/z) were collected under optimal conditions.
2.7. Ethical Approval
All animal procedures were performed according to the Hungarian Animal Protection Act Scientific Procedures (Government Regulation 243/1998). The study was approved by the Animal Welfare Committee of the University of Pécs. Ethics Committee approved the use of animals in this study under protocol number BA02/2000-21/2016 (Committee on Animal Research and Ethics, Department of Food Chain Safety, Plant Protection and Soil Conservation of the Baranya County Government, Pécs, Hungary).
3. RESULTS
3.1. HPLC-UV Method Validation
3.1.1. Specificity
Specificity was defined as the ability of the method to differentiate and quantify the analytes in the presence of endogenous constituents of the perfusate sample. (Fig. S1) depicts the representative HPLC chromatogram of the blank perfusate (with 3.5% PEG-400) (Fig. S1A and S1B) and the perfusate generated in rat small intestine luminal perfusion experiments at the 45th minute of the perfusion period (Fig. S2A and S2B). The perfusates do not give a detectable chromatographic peak at the retention time of 1 (tR=25.50 min), 2 (tR=13.35 min), or any newly formed metabolite.
3.1.2. Linearity
Linearity was studied by analysis of standard solutions of 1 and 2 of five different concentrations (0.0004; 0.004; 0.02; 0.04; 0.16 mg/mL and 0.005; 0.01; 0.05; 0.1; 0.25 mg/mL, respectively) using blank perfusate as solvent. Each solution had a known concentration (50 μg/mL) of 2,5-dihydroxybenzoic acid (2,5-DHB) as the internal standard. Data were obtained from three parallel injections of two independent weighings of the substances. Calibration curves were generated by plotting the theoretical concentrations against the peak areas related to the internal standard (ratio of the peak areas of the examined compound and the internal standard). The calibration functions for 1 and 2 were y = 108.6239x + 0.0990 (r2 = 0.9998) and y = 24.4638x + 0.0132 (r2 = 0.9999), respectively.
3.1.3. System Suitability
System suitability data and system precision were evaluated based on the chromatogram of the solutions containing (a) 0.02 mg/mL of 1, (b) 0.1 mg/mL of 2 and 2,5-DHB (50 μg/mL) as internal standard. Results were obtained from 5 parallel injections each. The evaluation was based on relative standard deviation (RSD%). System suitability data are summarized in Table 1. (Fig. S3A and S3B) show the representative HPLC chromatograms of system suitability.
3.1.4. Precision
Precision was studied by investigating repeatability and intermediate precision. Repeatability was determined by measuring intra-day data of three parallel injections of three parallel dilutions of two independent weighings of 1 and 2 at three different concentration levels (c = 0.16; 0.02; 0.004 mg/mL and c = 0.25; 0.05; 0.005 mg/mL, respectively). Repeatability data of compounds 1 and 2 are summarized in Tables 2 and 3, respectively.
Intermediate precision was determined by measuring inter-day (by injection of the samples over three consecutive days) data of three parallel injections of three dilutions from two independent weighings of 1 and 2 solutions at the three different concentration levels. The evaluation was based on relative standard deviation (RSD %). The respective precision data are summarized in Table S1 and S2. The data indicate that analysis of the samples provides acceptable (intra- and interday) repeatability and accuracy.
3.1.5. Accuracy
To determine accuracy, the average of the measured and the expected concentrations of 1 or 2 at three concentration levels (c = 0.08; 0.032; 0.012 mg/mL and c = 0.15; 0.075; 0.025 mg/mL, respectively) were compared. Measured concentrations were determined from 5 parallel injections of the solutions treated as described in item 2.3. Accuracy (recovery) data were calculated, expressing the measured concentrations as expected percentages. Measured concentrations were determined using the respective calibration equations 1 and 2. Data are summarized in Table 3S and Table 4S. The results indicate that the accuracy (recovery) data (%RSD) of 1 falls into a narrower region (1.17-1.23%) than those of 2 (0.38-2.43%). Both are within the ±5% range of the nominal concentrations.
| Compound/Method | tR | k' | Asym | N | Rs |
| 2,5-DHB/Method 1 | 3.20 | 0.53 | 0.65 | 3520 | |
| Compound 1/Method 1 | 25.40 | 11.04 | 0.82 | 182182 | 97.20 |
| 2,5-DHB/Method 2 | 5.50 | 1.6 | 0.66 | 1779 | |
| Compound 2/Method 2 | 13.35 | 5.29 | 0.61 | 66978 | 21.39 |
| c (mg/mL) | Weighing | Dilution | AI.S. | A1 | A1/AI.S. | c (mg/mL) | c (Mean) (mg/mL) |
| 0.16 | 1 | 1 | 912.6 | 17383.4 | 19.048 | 0.1745 | 0.1672 |
| 916.2 | 17652.3 | 19.267 | 0.1765 | ||||
| 920.7 | 15160.9 | 16.467 | 0.1507 | ||||
| 2 | 970.5 | 17448 | 17.978 | 0.1646 | 0.1628 | ||
| 969.2 | 17253.1 | 17.801 | 0.1630 | ||||
| 960.4 | 16859.2 | 17.554 | 0.1607 | ||||
| 2 | 3 | 989.6 | 16950.9 | 17.129 | 0.1568 | 0.1523 | |
| 999.2 | 16709.9 | 16.723 | 0.1530 | ||||
| 1003.1 | 16108.1 | 16.058 | 0.1469 | ||||
| Results* | Mean | 960.2 | 16836.2 | 17.559 | 0.1607 | ||
| SD | 35.7 | 781.0 | 1.101 | 0.0101 | |||
| %RSD | 3.7 | 4.6 | 6.270 | 6.3060 | |||
| c (mg/mL) | Weighing | Dilution | AI.S. | A1 | A1/AI.S. | c (mg/mL) |
c(Mean) (mg/mL) |
| 0.02 | 1 | 1 | 1040.8 | 2316.9 | 2.226 | 0.0196 | 0.0198 |
| 1036.6 | 2324.8 | 2.243 | 0.0197 | ||||
| 1034.1 | 2362.1 | 2.284 | 0.0201 | ||||
| 2 | 1031.2 | 2398.4 | 2.326 | 0.0205 | 0.0203 | ||
| 1030.2 | 2381.6 | 2.312 | 0.0204 | ||||
| 1033.7 | 2341.9 | 2.266 | 0.0199 | ||||
| 2 | 3 | 899.7 | 2092.5 | 2.326 | 0.0205 | 0.0199 | |
| 922.0 | 2093.7 | 2.271 | 0.0200 | ||||
| 934.0 | 2049.2 | 2.194 | 0.0193 | ||||
| Results* | Mean | 995.8 | 2262.3 | 2.272 | 0.0200 | ||
| SD | 58.7 | 140.8 | 0.046 | 0.0004 | |||
| %RSD | 5.9 | 6.2 | 2.006 | 2.0973 | |||
| c (mg/mL) | Weighing | Dilution | AI.S. | A1 | A1/AI.S. | c (mg/mL) |
c(Mean) (mg/mL) |
| 0.004 | 1 | 1 | 951.9 | 465.6 | 0.489 | 0.0036 | 0.0037 |
| 951.0 | 473.7 | 0.498 | 0.0037 | ||||
| 954.7 | 481.3 | 0.504 | 0.0037 | ||||
| 2 | 958.0 | 451.2 | 0.471 | 0.0034 | 0.0036 | ||
| 952.0 | 454.3 | 0.477 | 0.0035 | ||||
| 934.6 | 501.1 | 0.536 | 0.0040 | ||||
| 2 | 3 | 879.8 | 457.1 | 0.520 | 0.0039 | 0.0040 | |
| 852.9 | 455.5 | 0.534 | 0.0040 | ||||
| 851.3 | 452.6 | 0.532 | 0.0040 | ||||
| Results* | Mean | 920.7 | 465.8 | 0.507 | 0.0038 | ||
| SD | 45.7 | 16.8 | 0.025 | 0.0002 | |||
| %RSD | 5.0 | 3.6 | 4.903 | 6.0937 | |||
3.1.6. Determination of LOD and LOQ
The limit of detection (LOD) was determined experimentally and taken as the concentration producing a detector signal that could be clearly distinguished from the baseline noise (3 times baseline noise). The limit of quantitation (LOQ) was defined as the concentration that produced a detector signal ten times greater than the baseline noise [40]. The LOD value for 1 was 0.0002 mg/mL and that for 2 was 0.001 mg/mL. The LOQ value for 1 was 0.0005 mg/mL and that for 2 was 0.0025 mg/mL.
3.2. Analysis of the Perfusates
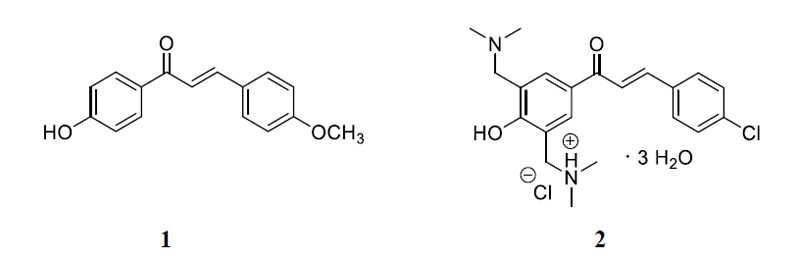
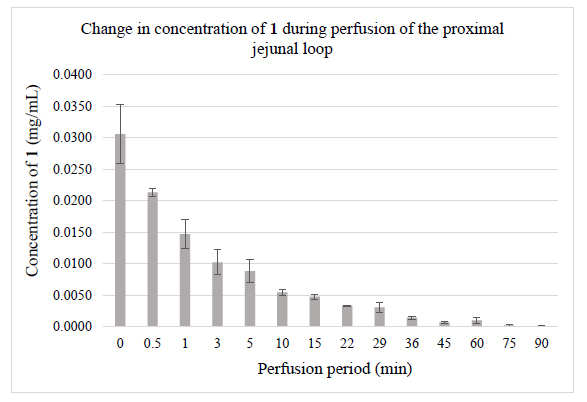
The validated HPLC-UV-Vis methods were applied for monitoring jejunal elimination of chalcones 1 and 2 in samples generated in rat small intestine luminal perfusion experiments. (Fig. 2) demonstrates the luminal elimination of chalcone 1 (Fig. 2A) and chalcone 2 (Fig. 2B) while the isotonic buffer solutions of the two compounds were perfused in the 90-minute experiments. Data represent the average of three parallel experiments. It was found that less polar 1 has a better penetration efficiency under the experimental conditions.
| c (mg/mL) | Weighing | Dilution | AI.S. | A2 | A2/AI.S. | c (mg/mL) |
c (Mean) (mg/mL) |
| 0.25 | 1 | 1 | 1417.2 | 10636.0 | 7.505 | 0.3062 | 0.3019 |
| 1423.3 | 10615.4 | 7.458 | 0.3043 | ||||
| 1440.3 | 10418.5 | 7.234 | 0.2951 | ||||
| 2 | 1525.7 | 9899.7 | 6.489 | 0.2647 | 0.2644 | ||
| 1519.1 | 9810.7 | 6.458 | 0.2634 | ||||
| 1530.7 | 9946.6 | 6.498 | 0.2651 | ||||
| 2 | 3 | 1487.2 | 10932.7 | 7.351 | 0.3000 | 0.2983 | |
| 1411.4 | 10192.4 | 7.221 | 0.2946 | ||||
| 1319.7 | 9711.2 | 7.359 | 0.3003 | ||||
| Results | Mean | 1452.7 | 10240.4 | 7.064 | 0.2882 | ||
| SD | 69.5 | 429.8 | 0.446 | 0.0182 | |||
| %RSD | 4.8 | 4.2 | 6.313 | 6.3252 | |||
| c (mg/mL) | Weighing | Dilution | AI.S. | A2 | A2/AI.S. | c (mg/mL) |
c (Mean) (mg/mL) |
| 0.05 | 1 | 1 | 1578.7 | 2055.1 | 1.302 | 0.0527 | 0.0526 |
| 1577 | 2048.4 | 1.299 | 0.0526 | ||||
| 1577.1 | 2045.9 | 1.297 | 0.0525 | ||||
| 2 | 1544.9 | 2264.7 | 1.466 | 0.0594 | 0.0581 | ||
| 1508.9 | 2105.6 | 1.395 | 0.0565 | ||||
| 1387.9 | 2000.8 | 1.442 | 0.0584 | ||||
| 2 | 3 | 1612.6 | 2173.9 | 1.348 | 0.0546 | 0.0560 | |
| 1514.7 | 2244.6 | 1.482 | 0.0600 | ||||
| 1673.5 | 2205.8 | 1.318 | 0.0533 | ||||
| Results* | Mean | 1552.8 | 2127.2 | 1.372 | 0.0555 | ||
| SD | 79.5 | 97.2 | 0.076 | 0.0031 | |||
| %RSD | 5.1 | 4.6 | 5.504 | 5.5576 | |||
| c (mg/mL) | Weighing | Dilution | AI.S. | A2 | A2/AI.S. | c (mg/mL) |
c (Mean) (mg/mL) |
| 0.005 | 1 | 1 | 1423.6 | 201.3 | 0.141 | 0.0052 | 0.0056 |
| 1425.2 | 223.4 | 0.157 | 0.0059 | ||||
| 1426.6 | 214.4 | 0.150 | 0.0056 | ||||
| 2 | 1593.2 | 221.9 | 0.139 | 0.0052 | 0.0052 | ||
| 1594.7 | 225.8 | 0.142 | 0.0052 | ||||
| 1594.1 | 223.3 | 0.140 | 0.0052 | ||||
| 2 | 3 | 1466.6 | 212.3 | 0.145 | 0.0054 | 0.0055 | |
| 1464.4 | 222.4 | 0.152 | 0.0057 | ||||
| 1464.1 | 214.2 | 0.146 | 0.0054 | ||||
| Results* | Mean | 1494.7 | 217.7 | 0.146 | 0.0054 | ||
| SD | 76.4 | 7.8 | 0.006 | 0.0002 | |||
| %RSD | 5.1 | 3.6 | 4.133 | 4.5441 | |||
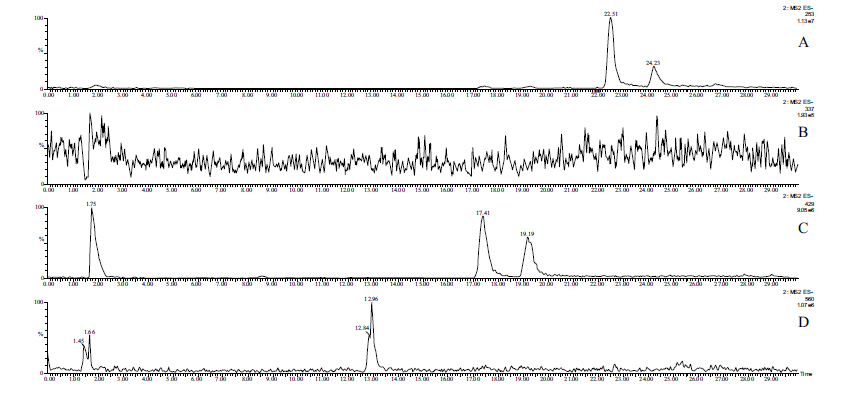
Due to the low concentrations, HPLC-MS methods combined with solid-phase extraction have been developed to detect and identify possible Phase 2 metabolites in the perfusates. The molecular ion chromatograms of the two perfusates are shown in (Figs. 3-5). In the case of chalcone 1, the HPLC-MS method with ESI(+) ionization provided separation and detection of the parent compound 1 (tR= 22.52 min and 24.24 min) as well as its sulfate- (tR=14.86 min) (1-SO3H), glucuronide- (tR=17.39 min and 19.18 min) (1-GLU), and GSH-conjugates (tR=12.90 and 13.79 min) (1-GSH) in the perfusate (Fig. 3). HPLC-MS analysis of the samples with ESI(-) ionization indicated the presence of only the parent compound (1) and its glucuronide (1-GLU) (Panel C) and GSH- (1-GSH) (Panel D) conjugates (Fig. 4). The structure of the formed metabolites is shown in Fig. (4S).
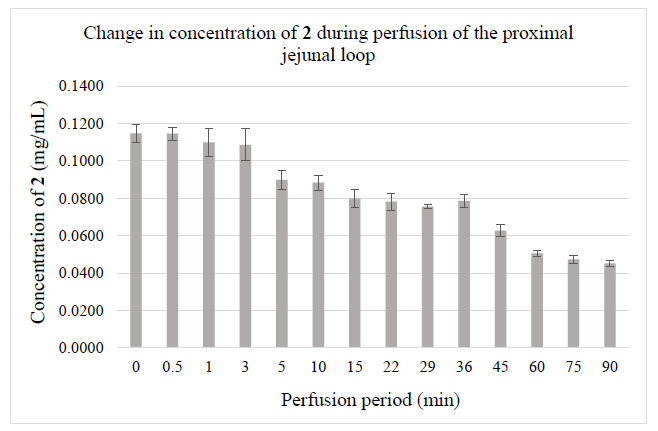
The parent compound 1 (Panel A), its glucuronide-(Panel C), and GSH-conjugates (Panel D) were identified as mixtures of diastereoisomers (Figs. 3 and 4). It is worth mentioning that the formed (Z)-isomer is not separated using the somewhat different method in the HPLC-UV analysis of the samples (Fig. S1B). The structures of the compounds with rather short retention times (Panels B and C) could not be identified. Results of the ESI (+) and ESI (-) HPLC-MS analyses of the perfusates are summarized in Table 4.
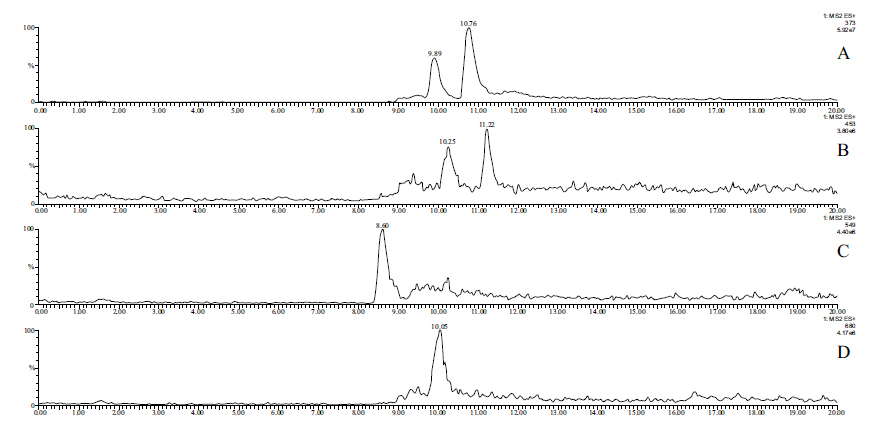
In the case of the bis-Mannich analog 2, HPLC-MS analysis of the perfusate with ESI (+) ionization provided separation and identification of the parent compound 2 (tR=9.89 min and 10.76 min) as well as its sulfate- (tR= 10.25 min and 11.22 min) (2-SO3H), glucuronide- (tR=.8.60. min) (2-GLU) and GSH-conjugates (tR= 10.05 min) (2-GSH). In the chromatograms, the parent compound (2) and its sulfate conjugate (2-SO3H) were identified as mixtures of diastereomers (Fig. 5). (Similar to 1, the (E) and (Z) isomers of 2 are not separated under the HPLC-UV conditions (Fig. S2B). The structure of the formed metabolites is shown in (Fig. 5S). Results of the ESI(+ mode) and ESI(- mode) HPLC-MS analysis of the perfusates are summarized in Table 5.
| Compound | Molar Mass |
tR (min) ESI (+) |
tR (min) ESI (-) |
| 1 | 254.09 | m/z 255 tR1=22.52; tR2=24.24 | m/z 253 tR1=22.51; tR2=24.23 |
| 1-SO3H | 334.05 | m/z 335 tR=14.86 | m/z 333 undetectable |
| 1-GLU | 430.13 | m/z 431 tR1=17.39; tR2=19.18 | m/z 429 tR1=17.41; tR2=19.19 |
| 1-GSH | 561.18 | m/z 562 tR1=12.90; tR2=13.79 | m/z 560 tR1=12.96; tR2=undetectable |
| Compound | Molar Mass (Base) |
tR (min) – ESI (+) |
tR (min) ESI (-) |
| 2 | 372.16 | m/z 373 tR1=9.89; tR2=10.76 | m/z 371 undetectable |
| 2-SO3H | 452.12 | m/z 453 tR1=10.25; tR2=11.22 | m/z 451 undetectable |
| 2-GLU | 548.19 | m/z 549 tR=8.60 | m/z 547 undetectable |
| 2-GSH | 679.24 | m/z 680 tR=10.05 | m/z 678 undetectabble |
4. DISCUSSION
The absorption and metabolism of two 4’-hydroxychalcone derivatives were investigated by an ex vivo rat model [34, 38, 39]. The intestinal perfusion model is based on the principle that drug concentration in perfusion solution decreases over time, attributed to the net drug absorption. Chalcones 1 and 2 were luminally perfused in the small intestine at a concentration of 240 µM and 280 μM, respectively, and elimination of the compounds was determined by HPLC-UV analysis of the perfusion solutions.
Our results show that over the 90 min experiments, almost the total amount of chalcone 1 was eliminated from the perfusate following a quadratic function (y=0.0003x2-0.0064x +0.0341; r2=0.9295), while the bis-Mannich analog 2 appears in approximately half of the initial tested concentration following a linear equation (y=-0.0067x+0.1469; r2=0.9933) (Figs. 2A and 2B). Several studies have highlighted that careful consideration should be given to the ionization profiles of compounds in predicting their bioavailability [41-44]. The pKa value of the dimethylamino substituent of 2 is high [45]; it is, therefore, predominantly ionized in the gastrointestinal tract. Conversely, the high pKa of the hydroxyl phenolic groups of 1 renders it predominantly uncharged at physiological pH, making its permeability possible through passive diffusion [46]. However, concerning chalcone 2, due to the high degree of ionization, the passage of these molecules across the membrane requires the activity of specific transport and channel proteins [19, 20].
On the other hand, glucuronidation stability of Mannich derivatives of the flavonoid baicalein has been attributed to the intramolecular hydrogen bond between the lone pair of the dimethylamino group with its adjacent phenolic group, reducing the nucleophilic characteristics of the hydroxyl group [47]. A similar intramolecular hydrogen bond of 2 was detected by crystallographic characterization of the compound in our previous study [30]. Such resistance of glucuronide formation can also hinder intestinal absorption of 2 compared to 1.
The metabolic activity of the intestinal lumen is well documented. The main metabolic enzymes reported to be expressed in the rat enterocytes are CYP1A1, 2B1, and 3A1 [48, 49], GST [50], SULT [51, 52], and UGT1A forms [53]. MRP2 is localized to the apical membrane domain of polarized cells such as intestinal epithelia, where it mediates unidirectional transport of substrates to the luminal side of the organ [54]. Recently, we have demonstrated absorption of capsaicin [34] and 4-nitrophenol [38, 39] from rat small intestine and luminal excretion of their glucuronide conjugates. On the contrary, no glucuronide conjugate of ibuprofen could be detected in the luminal perfusate under the same experimental conditions [55]. Such observation can result from a lack of expression of the UGTA2B1 isoform in the rat small intestine epithelial cells [56]. Accordingly, it cannot be excluded that the difference in metabolic characteristics of the two chalcones also plays some role in their different intestinal elimination rates.
Based on the above previous results, HPLC MS analysis of the intestinal perfusates was performed to investigate the presence of possible Phase 2 metabolites of the two chalcones. After solid-phase extraction, the expected (glucuronide-, sulfate- and glutathione (GSH)-) conjugates were detected and identified by HPLC-ESI-MS in positive and negative modes. According to these analyses, the parent compounds (1 and 2) gave two peaks with an identical molecular weights indicating (E)/(Z) isomerization under the experimental conditions. Light-initiated (E)/(Z) isomerization is a well-documented property of chalcones [57]. On the other hand, reversible thia-Michael addition of cellular thiols (e.g., GSH) also results in the formation of (Z) isomers of chalcones [58].
Similarly, two chromatographic peaks with identical molecular mass could be detected for the 1-GLU and the 2-SO3H conjugates (Fig. 3-5). Further studies are needed to clarify the stereochemistry of the diastereomeric structures.
GSH-conjugation is a well-known metabolic transformation of chalcones and other enones by Michael-type addition reactions. In these reactions, the nucleophilic sulfur is added to the enone moiety's beta-carbon atom, generating a new chiral center. Due to the chiral centers inherent in GSH, the formation of the enantiomeric forms of the new chiral center results in the formation of two diastereomeric adducts. Contrary to both identified isomers of 1-GSH, only one of the two possible diastereomeric adducts of 2-GSH could be detected. However, chalcone-GSH adducts can be separated only under optimized chromatographic conditions [58]. The results showed that metabolites from chalcone 2 conferred predominant single-charged protonated precursor [M+H]+ in the positive ion mode (ESI+).
CONCLUSION
Utilizing validated HPLC-UV-Vis methods, it was demonstrated that elimination of the bis-Mannich derivative 2 is slower from the rat small intestine than that of the analogous 4’-hydroxychalcone 1. The absorbed chalcones undergo Phase 2 metabolic transformations in the intestinal epithelial cells, and the glucuronide, the sulfate, and the glutathione conjugates of the parent compounds appear in the intestinal perfusate. HPLC-MS method combined with solid-phase extraction allowed structural identification of the conjugates. The results demonstrate, to the best of our knowledge, for the first time, how such structural modification of phenolic chalcones affects intestinal elimination and metabolism of the compounds.
LIST OF ABBREVIATION
| PEG | = Polyethylene Glycol |
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
The study was approved by the Animal Welfare Committee of the University of Pécs. Ethics Committee approved the use of animals in this study under protocol number BA02/2000-21/2016 (Committee on Animal Research and Ethics, Department of Food Chain Safety, Plant Protection and Soil Conservation of the Baranya County Government, Pécs, Hungary).
HUMAN AND ANIMAL RIGHTS
No humans were used for studies that are the basis of this research. All the animals used were in accordance with Hungarian Animal Protection Act Scientific Procedures (Government Regulation 243/1998).
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
The data sets used and/or analysed during this study are available from the corresponding author upon request.
FUNDING
The authors express their sincere thanks to the EFOP Operational Program (Grant No. EFOP-3.6.1-16-2016-00004) and the Conselho Nacional de Desenvolvimento Científico Tecnológico (CNPq) (Grant No. 478337/2013-2) for providing financial support.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
ACKNOWLEDGEMENTS
The authors express their special thanks to Coordenadoria de Aperfeiçoamento de Pessoal de Ensino Superior (CAPES) (Grant No. BEX 6234/15-1) for a fellowship (to A.B.) for supporting this work. The authors express their sincere thanks to the EFOP Operational Program (Grant No. EFOP-3.6.1-16-2016-00004) and the Conselho Nacional de Desenvolvimento Científico Tecnológico (CNPq) (Grant No. 478337/2013-2) for providing financial support.
SUPPLEMENTARY MATERIAL
Supplementary material is available on the publisher’s website along with the published article.

