All published articles of this journal are available on ScienceDirect.

Acridine and Acridinones: Old and New Structures with Antimalarial Activity
Abstract
Since emergence of chloroquine-resistant Plasmodium falciparum and reports of parasite resistance to alternative drugs, there has been renewed interest in the antimalarial activity of acridines and their congeners, the acridinones. This article presents literature compilation of natural acridinone alkaloids and synthetic 9-substituted acridines, acridinediones, haloalcoxyacridinones and 10-N-substituted acridinones with antimalarial activity. The review also provides an outlook to antimalarial modes of action of some described compounds.
INTRODUCTION
Malaria is the most important parasitic disease in the world. Its aetiological agents are protozoa of the genus Plasmodium. P. falciparum is the most virulent among the four species infecting humans and is responsible for most of mortality. In 2008, among 3.3 billion people at risk , there were 243 million malaria cases, causing an estimated 863 000 deaths, mostly of children under five years. From 109 countries endemic for malaria, 45 were within the WHO (World Health Organization) African Region [1].
The combination of tools and methods to combat malaria now includes only artemisinin-based combination therapy (ACT) to treat this infectious disease [1]. Resistance of P. falciparum has emerged to all classes of antimalarial drugs except artemisinin. Although resistance to artemisinin derivatives has not yet been identified, with increased use of these drugs, it is expected that resistant parasites will be selected.
The lack of structural diversity of currently used antimalarials is evident. A small number of chemically related drugs belonging to five classes of compounds: 4-aminoquinolines (chloroquine (CQ), quinine, mefloquine, amodiaquine and halofantrine), 8-aminoquinolines (primaquine), antifolate compounds, atovaquone and artemisinin derivatives are currently in use [2]. Is obvious the urgent need for new antimalarials.
Acridine and acridinone are chemical families differing from most current antimalarials. Several derivatives have demonstrated strong antimalarial activity; two of them have been clinically tested.
Quinacrine (QA), 6-chloro-2-methoxy-9-(4-diethylamino-1-methylbuthylamino)acridine (Fig. 1), was the first known clinically tested antimalarial. This 9-aminoacridine was discovered in 1933 following an intensive search for synthetic quinine substitutes and was used when quinine became practically unavailable during World War II [3]. Although it is not currently in use as antimalarial, this drug has inspired many scientists to design, synthesise and evaluate many related derivatives.
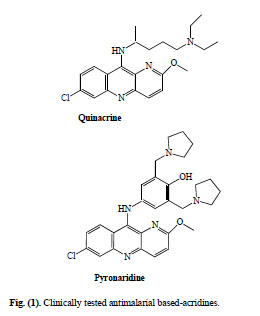
Clinically tested antimalarial based-acridines.
Pyronaridine (Pyr), (2-methoxy-7-chloro-10-[3´,5´-bis(pyrrolidinyl-1-methyl) 4´hydroxyphenyl] aminobenzyl-(b)-1,5-naphthyridine) (Fig. 1), the other clinically tested acridine-based antimalarial, was designed and synthesised at the Institute of Parasitic Diseases, Shanghai, in 1970. Clinical trials began in 1971 and nine years later was formally released as a new antimalarial drug in China [4]. Now is being used as fixed-dose combination with artesunate (PYRAMAX®), for uncomplicated P. falciparum and P. vivax malaria as a once a day for three days treatment [5]. In 2008, four pivotal Phase III trials were completed; the paediatric granular formulation were registered in 2009 (http://www.mmv.org).
Recent structure-activity relationship studies have revealed new promising antimalarials based on acridine and acridinone core. Haloalcoxyacridinones, extremmely potent antiplasmodial activity in vitro [6], novel acridinedione with potent inhibitory activity of mitochondrial bc1 Complex [7], and 10-N-substituted acridinones showing strong chemosensitizing properties and heme polymerisation inhibition [8] are the most significant structures.
This review covers the antimalarial acridine and acridinones from natural and synthetic sources. Information on mechanisms of antimalarial activity is also discussed.
NATURAL STRUCTURES
Acridinone alkaloids are synthesised by plants of Rutaceae family. Most of these structures have exhibited high activity against tumoral cell lines and some of them have been tested against malaria parasites.
Acronycine (1, Fig. 2) was first isolated from the Australian scrub ash Acronychia baueri, it showed antineoplastic [9] and antiparasitic activity [10]. The antimalarial activity was firstly tested in vitro against Plasmodium yoelii [11]. Acronycine and 5-hydroxyacronycine (2, Fig. 2) suppressed almost 90 % of parasitemia in vitro, however, they were moderately active against P. falciparum [12]. Furthermore, many derivatives of this alkaloid were evaluated, only 2-nitroacronycine(3, Fig. 2) improved the antimalarial activity, reaching IC50 values around 2 μg/mL for susceptible and resistant strains [12].
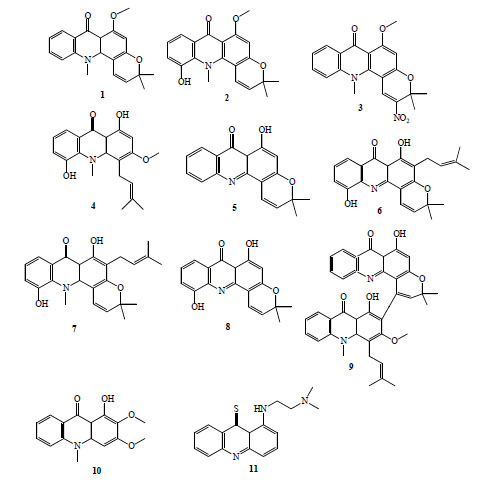
Antimalarial acridinone alkaloids (compounds 1-11).
Other seven kinds of acridinone alkaloids obtained from Rutaceae plants: glycocitrine-I (4), des-N-methylnor-acronycine (5), atalaphillinine (6), 5-hydroxy-N-methylseverifoline (7), atalaphillidine (8) and glycobismine-A (9), Fig. (2), exhibited antimalarial activity comparable to chloroquine diphosphate using the model of P. yoelii described by Fujioka et al. [11]. Atalaphillinine inhibited completely the development of malaria parasites in vivo after prophylactic administration in a daily dose of 50 mg/Kg for 3 days with no obvious toxic effect [11]. Forty-seven new acridinone alkaloid derivatives were tested for these authors. Four compounds exhibited potent antimalarial activity (IC50 of 23-150 ng/mL) against P. yoelii in vitro [13].
Arborinine (10, Fig. 2) and normelicopicine, acridinone alkaloids isolated from Teclea thiocarpa (Rutaceae: Toddalioideae), posses modest antiplasmodial activity against P. falciparum [14]. Normelicopicine caused 32 % parasitemia suppression at a dose of 25 mg/Kg [14].
Acronycine-inspired thioacridinones were synthesised for evaluation as antitumor agents. The thioacridinones were also tested for antimalarial activity against a CQ-sensitive strain of P. falciparum; all products showed antiplasmodial activity (IC50 0.4-27 μg/ml). The most potent compound, 8-{[2-(dimethylamino)ethyl] amino}acridine-9(8aH)-thione), (11, Fig. 2) also effectively inhibited the resistant strain growth [15].
SYNTHETIC ACRIDINES
9-Aminoacridines
A strategy for optimising the 9-aminoacridine antimalarial activity has been recently used, that involves the synthesis of 6-chloro-2-methoxyacridine derivatives [16-18].
Winter et al. [6] reported strong in vitro antimalarial activity for 2-methoxy-6-chloro-9 aminoacridine (12, Fig. 3) (IC50 = 18-42 nM) and 3-(6,6,6-trifluorohexyloxy)-6-chloro9-aminoacridine (13, Fig. 3) (IC50 = 2.9-10 nM), however, moderate IVTI (in vitro therapeutic indexes), lower than 100, were developed using murine splenic lymphocytes.
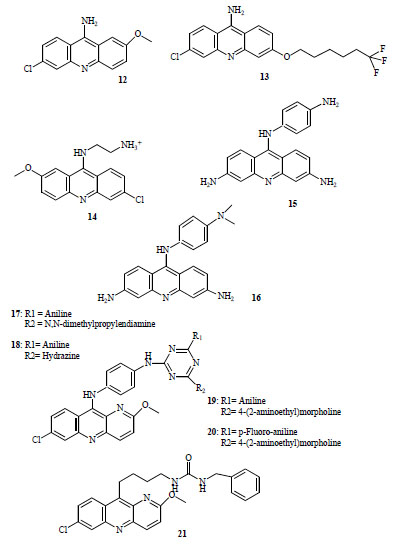
Antimalarial acridines (compounds 12-21).
Series of 9-aminoacridine derivatives showed significant antimalarial activity against CQ-resistant strains with IC50 values ≤ 0.2 μM [18]. More recently, these authors have synthesized new molecules containing the same scaffold [19]. In vitro antimalarial evaluation showed that two positive charges as well as the 6-chloro and 2-methoxy substituents on the acridine ring were required to exert good antimalarial activity. Two compounds inhibited the growth of CQ-susceptible strain with IC50 ≤ 0.07 μM, the IC50 value was lower than CQ for resistant strains. 9-(6-ammonioethyl-amino)-6-chloro-2-methoxyacridinium dichloride(14, Fig. 3) was the most promising agent displaying the lowest toxicity against KB cells line, exhibiting moderate selectivity indexes ranging from 7 for CQ-resistant strains to 50 for susceptible strain.
9-Anilinoacridines
These acridine derivatives have well described biological actions and many have been used successfully to treat different forms of leukemia [20]. Following the principle of a common intracellular target (Topoisomerase II), a series of 9-anilinoacridines were synthesized with varied acridine ring and anilino substituents which showed difference in structure-activity relationship against P. falciparum [21-23]. Most active compounds possess 3,6 diamino substitutions on the acridine ring (3,6-diamino-1’-amino-9-Anilinoacridine (15, Fig. 3), 1’-dimethylamino-3,6-diamino-9-Anilinoacridine (16, Fig. 3)) exhibiting IC50 values in the low nanomolar range against K1 (CQ-resistant strain) [22, 24]. The overall hydrophilicity and the high basicity are important determinants of parasite inhibition. This dependence on hydrophilic character seems reflect drug penetration factors [22].
Recently, antimalarial evaluation of a series of hybrid 9-anilinoacridine/ triazine was reported [25]. Two compounds, N2-{4-[(7-chloro-2-methoxy-10,10a-dihydrobenzo[b]-1,5-naphthyridin-10-yl)amino]phenyl}-N4-1-dimethylamino) propyl]-N6-phenyl-1,3,5-triazine-2,4,6-triamine (17, Fig. 3) and N-{6-[(7-chloro-2-methoxy-10,10a-dihydrobenzo-[b]-1,5-naphthyridin-10-yl)amino]pyridin-3-yl}-6-hydrazino-N'-phenyl-1,3,5-triazine-2,4-diamine (18, Fig. 3) displayed two times higher potency than CQ, against a sensitive-strain (IC50 around 4.2 nM) and fairly high selectivity index (SI > 295) respect to VERO cells cytotoxicity. However, no significant protection by oral administration of 100 mg/Kg of active compounds was obtained in 28 days survival assay using mice infected with CQ-resistant N-67 strain of P. yoelii. Other two compounds, N2-{4-[(7-chloro-2-methoxy-10,10a-dihydrobenzo[b]-1,5-naphthyridin-10-yl)-amino]phenyl}-N4-(2-morpholin-4-ylethyl)-N6-phenyl-1,3,5-triazine-2,4,6-triamine (19, Fig. 3) and -N2-{4-[(7-chloro-2-methoxy-10,10a-dihydrobenzo[b]-1,5-naphthyri-din-10-yl)amino]phenyl}-N4-(4-fluorophenyl)-N6-(2-mor-pholin-4-ylethyl)-1,3,5-triazine-2,4,6-triamine (20, Fig. 3), less active in vitro, displayed more than 95 % suppression in the same in vivo model.
Other 9-Substituted Acridines
Sulfonamide and urea derivatives of QA with varying methylen spacer lengths have been tested for in vitro activity against CQ-sensitive and resistant strains [26]. One urea derivative, N-benzyl-N'-[4-(7-chloro-2-methoxybenzo[b]-1,5-naphthyridin-10-yl)butyl]urea (21, Fig. 3), showed potent activity against the sensitive strain although low selectivity index was obtained.
Aryl sulfones and aryl thioacridine were studied in an approach to obtain combined functions, QA and sulfonamide or sulfone drugs targets, in the same structure [27]. IC50 values are included from 4 up to > 500 μM for susceptible strain and from 6.4 up to > 500 μM for resistant strain. No improvement of antimalarial activity was observed.
SYNTHETIC ACRIDINONES
Acridinediones
Floxacrine, 7-chloro-10-hydroxy-3-[4-(trifluoromethyl)-phenyl]-3,4-dihydroacridine-1,9(2H,10H)-dione (22, Fig. 4), is one of a group of dihydroacridinediones, originally synthesized searching for novel and effective antibacterial drugs [28]. Significant prophylactic antimalarial activity was exhibited by this compound [29], but unfortunately, it had several liabilities, including limited water solubility [30], parasite drug resistance [28, 31] and dose dependent periarteritis [31].
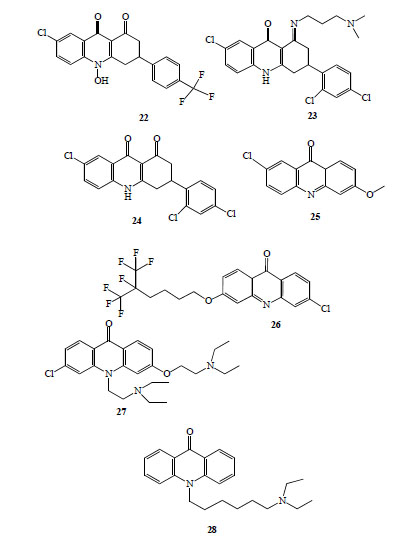
Antimalarial acridinones (compounds 22-28).
Those reasons led to substantial efforts to identify an analogue of floxacrine without those drawbacks [30, 32-34]. A QA-Floxacrine, hybrid, WR 243251,(1E)-7-chloro-3-(2,4-dichlorophenyl)-1-{[3-(dimethylamino)propyl]imino}-1,3,4,10-tetrahydroacridin-9(2H)-one (23, Fig. 4) [30, 32], devoid of the vascular cardiotoxicity of floxacrine, emerged as potential antimalarial agent.
Recently, other derivatives as WR 249685, 7-chloro-3-(2,4-dichlorophenyl)-3,4-dihydroacridine-1,9(2H,10H)-di-one (24, Fig. 4), WR 243246S-enantiomer, have shown strong antimalarial activity (IC50 ~ 15 nM) and specific inhibitory activity of parasite mitochondrial bc1 Complex [7].
Haloalcoxyacridinones
A novel class of highly active and selective antimalarial acridinones (haloalcoxyacridinones) was discovered using an empirical design strategy [6]. Firstly, the authors obtained 2-methoxy-6-chloroacridinone (25, Fig. 4) by mild hydrolysis of QA, this compound showed strong in vitro antimalarial activity (similar to 2-methoxy-6-chloro-9-aminoacridine (12, Fig. 3), IC50 of 45 nM for susceptible strain (D6) and 65 nM for multidrug resistant strain (Dd2), with higher SI). This acridinone was selected as lead to synthesized new analogues to develop a structure activity profile. Thirty compounds were evaluated, 11 acridinones showed IC50 values lower than CQ against susceptible (D6) and resistant (Dd2) P. falciparum strains. The most potent compounds share a unique and defining structural element in the C ring composed of an extended alkyl group terminated by one or more trifluoromethyl groups at the 3 position of the tricyclic acridone. 3-(5,6,6,6-Tetrafluoro-5-trifluoromethyl-hexyloxy)-6-chloroacridinone (26, Fig. 4) showed IC50 values ≈ 1 pM against the two strains and IVTI higher than 100 000 [6].
10-N-Substituted Acridinones
New interesting acridinones were synthesised and tested [35]. Alkyl side chains (with lengths ranging from two to eight carbons) with tertiary amino group at the terminal position were attached at N 10 position of acridinones. Those compounds exhibited antimalarial in vitro activity in the µM range but display CQ-chemosensitizing activities against Dd2 strain. More recently, these authors [8] designed, synthesized and tested a dual function acridinones, incorporating into one molecule a heme- targeting tricyclic mainframe with an ionoizable side chain to promote accumulation in the digestive vacuole and a chemosensitization moiety at the N 10 position. The lead compound, T3.5, (3-chloro-6-(2-diethylamino-ethoxy)-10-(2-diethylamino-ethyl)-acridino-ne), (27, Fig. 4), showed IC50 values in the nM range against a panel of CQ-susceptible and resistant strains and proved synergistic with chloroquine, amodiaquine (verapamil-like chemosensitization) and quinine or piperaquine against Dd2 strain. In vivo evaluation, using murine malaria parasites (P. yoelii and P. berghei) demonstrated a curative action of 256 mg/Kg/day orally or 200 mg/Kg/day intraperitoneally. No overt toxicity or behaviour change was observed in animals.
MECHANISMS OF ANTIMALARIAL ACTIVITY
In order to design and develop new therapies that circumvent resistance many efforts are dedicated to elucidate the mode of action of new synthesised compounds and clinically tested agents which mechanism is not completely clarified, as aminoquinoline and artemisin derivatives [36].
Various mechanisms have been proposed for acridines and acridinones with antimalarial action. Inhibition of Hemozoin (β-hematin) formation, mitochondrial bc1 Complex and DNA Topoisomerase II, and interaction with DNA are the widest studied. In addition, acridinone core has been recently used to build potent chemosensitizers.
Inhibition of Hemozoin (β-Hematin) Formation
Malaria parasite survival is dependant on hemoglobin proteolysis. The amino acids derived from globin hydrolysis are incorporated into parasite proteins and appear to be available also for energy metabolism. Heme moiety is released as result of hemoglobin digestion. Lacking of heme oxygenase, plasmodial species sequester this toxic by-product into a chemically inert crystal. The product formed, hemozoin, is an aggregate of several units of hematin linked by carboxylate-iron(III) and carboxylate-carboxylate coordinated bonds. The aggregation process is a mechanism of detoxification that has been used as a target for antimalarial therapy [37]. Considerable data support the hypothesis that antimalarial quinolines inhibit parasite growth by binding to hematin preventing its agregation into the form of hemozoin [38].
Quinolines such as CQ form complexes with hematin in solution forming “π-π” or stacked complexes. Molecular planes are approximately parallel and in close proximity. Hydrophobic effect has a major influence on the free energy of complex formation. But, at the same time electrostatic and dispersion interactions are likely important as in other aromatic stacking interaction [38]. A previous study postulates that electronic features rather than steric factors seem to control primarily the inhibitory activity against hematin aggregation, concurring to a potential antimalarial activity [39]. Antimalarial compounds stabilize the dimer due to the wide range interactions determined by their complementary electrostatic profiles avoiding a further aggregation of this unit to form hemozoin. The positive potential of the aromatic ring of the active compounds would interact with negative potential on the center of the dimer. The regions of negative potential for the active compounds would stabilize the positive potential peripheral to the tetrapyrrol system of the hematin complex [39].
QA exhibited IC50 of 41 μM testing its action in β-hematin formation [40]. Other related structures with antimalarial activity have been also tested. Pyr (Fig. 1) targets hematin, as demonstrated by its ability to inhibit in vitro β-hematin formation (at a concentration equal to that CQ) and to form a complex with hematin with stoichiometry of 1:2 [41].
A series of 9-anilinoacridines were investigated for their abilities to inhibit β-hematin formation and to form drug hematin complexes [24]. In general, poor relation between inhibition and antimalarial activity was observed, only one compound (16, Fig. 3) may exerts its antimalarial activity by inhibiting hemozoin formation.
WR 243251(23, Fig. 4) was potent inhibitor of hematin polymerization (IC50 = 90 μM, CQ IC50 = 80 μM), however, this inhibition did not completely account for the increased antimalarial potency versus CQ for resistant strain K1 (IC50 = 9.5 nM, CQ IC50 = 450 nM) [42]. Floxacrine (22, Fig. 4) was 2-fold less potent than CQ for hematin polymerization inhibition and its analog WR 243246 (24, Fig. 4) was without activity indicating that it exerts antimalarial activity by other mechanisms [42].
Recently, the affinity of new dihydroacridinediones for heme and their ability to inhibit heme crystalization relative to known heme-binding drugs were determined [7]. In this study, Floxacrine (22, Fig. 4) inhibited heme crystalization with similar IC50 value that CQ whereas WR 249685 (24, Fig. 4) exhibited two-fold lower activity. Heme-drug equilibrium constants were calculated to be; 1.38 μM for CQ, 1.87 μM for Floxacrine (22, Fig. 4) and 31.7 μM for WR 249685. The relative poor heme binding affinity of WR 249685 (22, Fig. 4) was in contrast to its potent in vitro antimalarial activity (IC50 15 nM), this result suggest a different mechanism to exert the antimalarial action.
The dual function acridinone T3.5 (27, Fig. 4) interacts with heme to form a soluble complex with strong affinity at pH 5.2. This compound interferes with hemozoin, decreasing intracellular pigment incorporated during 24 hours per parasitized erythrocyte with susceptible and resistant strains. Visual localization of acridinone inside the P. falciparum-infected erythrocyte by confocal fluorescence microscopy indicates uptake and accumulation within the digestive vacuole [8].
Few studies about structure-inhibitory activity relationship for acridine and acridinone derivatives have been done. For aminoquinolines the group at the 7-position of the quinoline ring appears to be crucial in determining the ability to inhibit hemozoin formation, the presence of a chlorine atom at this position is an indicative of activity (43). QA (Fig. 1), Pyr (Fig. 1) and the dual function acridinone T3.5 (27, Fig. 4) contain the related chlorine group. In addition, the aminoalkyl side chain plays a relevant role on quinoline inhibitory activity [44]. QA (Fig. 1), Pyr (Fig. 1) and WR243251 (23, Fig. 4) possess an aminoalkyl side chain also, in contrast, low inhibition was observed testing WR 243246 (24, Fig. 4) [42] and its S-enantiomer WR 249685 (24, Fig. 4) [7] which do not contain that moiety. Tricycle mainframe with an ionizable side chain were designed to guaranteed heme-targeting of dual function acridinones, promoting accumulation in the digestive vacuole [8].
Inhibition of Mitochondrial bc1 Complex
In most eukaryotic cells, the mitochondrial electron transport chain (mtETC) is required for generation of the proton motive force, which is central to oxidative energy metabolism. However, in organisms or under conditions favoring a glycolytic metabolism, which include the blood stages of malaria parasite, the role of the mtETC is often reduced, and mitochondria may even become consumers rather than producers of energy (reviewed in Reference 45). Despite the relatively low activity of the mtETC in malaria parasites it still appears to be the primary source of the mitochondrial proton electrochemical gradient. Furthermore the sensitivity of parasites to inhibitors of the mtETC indicates that it is indispensable to the parasites [45].
The bc1 Complex (mitochondria Complex III) is a membrane-bound enzyme catalyzing the transfer of electrons from ubiquinol to cytochrome c coupled with the concomitant vectorial translocation of protons across the inner mitochondrial membrane [46]. The catalytic core of the enzyme is made up cytochrome b, cytochrome c1 and the Ruske iron-sulfur protein (ISP). The catalytic mechanism, known as the Q-cycle [46, 47] involves two distinct quinone binding sites within cytochrome b, the quinol oxidation site Qo and the quinone reduction site Qi [46]. These two sites are situated on opposite sides of the membrane linked by transmembrane electron pathway via hemes b1 and bh. A number of inhibitors selective for bc1Qo and Qi sites have been developed to control human pathogens.
Antimalarial drug atovaquone targets the Qo of cytochrome b in Plasmodium and related parasites [48]. WR 243251 (23, Fig. 4) exhibited cross resistance with atovaquone [49]. It reduced the rate of parasite oxygen consumption by almost 80% in the atovaquone-sensitive isolate and by 57 % in the atovaquone-resistant isolate. Drug interaction studies suggest that atovaquone and WR 243251 (23, Fig. 4) may inhibit growth additively or with mild synergy. Together these results suggest that while WR 243251 (23, Fig. 4) inhibits respiration, its mitochondrial target probably differ from that of atovaquone, besides, haem polymerization inhibition could be a further mechanism of action for this compound (see above).
Biagini et al. [7] investigated the antimalarial mode of action of other two different dihydroacridinediones, Floxacrine (22, Fig. 4) and WR 249685 (24, Fig. 4). Both of these compounds showed heme binding and bc1 inhibitory properties. However, whereas Floxacrine (22, Fig.4) exhibited the most potent heme polymerization inhibition, WR 249685 (24, Fig.4) is shown to be a highly species selective inhibitor of the Qo at the P. falciparumbc1. Last acridinedione displayed a Ki for P. falciparum of 0.3 nM and in vitro therapeutic index against human bc1 of > 4600. Docking of WR 249685 (24, Fig.4) to the P. falciparumbc1 model was energetically favourable, the model demonstrated selectivity in the docking of traditional Qo and Qi inhibitors. The most stricking feature of the model for dihydroacridinedione was the putative association between the inhibitor and the E-ef linker region of the cytochrome, a region of low sequence identity between P. falciparum and mammalian cytochrome b.
Interaction with DNA
Planar structure of tricycle rings confers to derivatives the ability to intercalate in the DNA and interfere with various metabolic processes both in procaryotic and eukaryotic cells.
This is the proposed mechanism of antitumoral activity for some acronycine analogues. The most potent antimalarial thioacridone also binds to DNA but no correlation seems to be apparent between IC50 value and DNA binding constant [50].
Depending on the nature of a particular compound and its substituents, the results and cellular consequences of acridine-DNA interaction may vary significantly [51, 52].
9-aminoacridines based structures (including QA, Fig. 1) are DNA-binding small molecules that do not induce DNA damage [53]. The interaction of QA (Fig. 1) with DNA was firstly reported by Van Dyke et al. [54]. They observed yellow-green fluorescence in nuclei of parasite P. falciparum in the presence of that acridine. In the course of mechanistic studies of QA (Fig. 1), it was noted that no inmediate connection between DNA interaction and therapeutic capacities was stablished [53]. New mechanisms have been discovered for QA (Fig. 1) and 9-aminoacridine derivatives in cancer cells. This compounds inhibited NFk B-mediated transcription and HSF1-mediated HSR [54, 55]. In the last study the authors hypothesised that the drugs may be effective in malaria inhibiting HSP synthesis, thereby sensitizing P. falciparum to fever induced by the host (e.g., heat shock) [55].
Antimalarial 9-anilinoacridines developed different ranges of DNA binding, Pyr (Fig. 1) also binds to DNA showing a concentration of 0.17 μM to displace 50 % of the bound ethidium from calf thymus DNA. In general, no correlation with parasiticidal action was observed [22]. Available evidences support a model whereby the acridine moiety intercalates into DNA and the 9-anilino side group projects into the DNA minor groove where it interacts with topoisomerases in cancer cells. The most widely and successfully used anticancer agents are the nonspecific DNA-damaging chemicals, including topoisomerases inhibitors [53]. Amsacrine (antitumoral acridine with moderate antimalarial activity) inhibit Topoisomerase II activity and alkylate DNA providing DNA damage [53].
Topoisomerase II Inhibition
Topoisomerases are essential for the modification of topology of nucleic acids. DNA topoisomerases are divided into two types: type I enzyme breaks one DNA strand for the passage of a second strand and the type II enzyme breaks both strands of one DNA duplex for the passage of a second DNA double strand [56].
The antimalarial compound 15 (Fig. 3) and the drug Pyr (Fig. 1) inhibited the decatenation activity of P. falciparum DNA topoisomerase II at concentration of 10 and 11 µM, respectively [22].
DNA cleavage is a transesterification reaction, and a covalent protein-DNA intermediate is formed during the transient DNA cleavage stage. This covalent topoisomerse II-DNA complex can be trapped in vitro by the addition of a protein denaturing such as sodium duodecyl sulphate (SDS) and is termed a cleavable complex [56].
The ability to generate in situ enzyme-DNA adducts or cleavable complexes by series of 9-anilinoacridines with antimalarial activity were examined [56]. 9-anilinoacridine derivatives with 3,6-diamino substituents produced protein-DNA complexes, whereas, Pyr (Fig. 1) did not generate it.
Chemosensitization
Chemosensitizers are compounds capable to reverse resistance, they lower the IC50 values of drugs against resistant parasites strains. Verapamil, a calcium channel blocker, was the first P. falciparum CQ-resistant reversal agent discovered [57]. Since then, many structurally and functionally diverse compounds have been identified and reported to demonstrate chemosensitization activity against malaria parasite, with antihistamines (e.g., chlorfeniramine) and tricyclic antidepressants (e.g., desipramine) among the most effective and best studied [58, 59].
CQ is a diprotic weak base, neutral and protonated species proportion varyies with pH. The neutral species enters the parasite and its internal compartments via simple difussion. Acidic enviroment of parasite´s digestive vacuole, increase the proportion of diprotonated CQ, which is unable to diffuse across the membrane and becomes trapped [60]. A recent study [60] demonstrated that chloroquine resistance is due to direct transport of the drug, out of the vacuole, and thus away from its site of action, via mutant CQ resistance transporter (PfCRT).
Mutations in the PfCRT protein, particularly the replacement of a lysine (K) with a threonine (T) at position 76, may play key roles in the mode of action of verapamil [61]. This K76T mutation occurs in a region of the protein that is predicted to be involved in substrate recognition [62]. The presence of a positive charge in the PfCRT substrate-binding site prevents protonated-CQ from interacting with the transporter [60]. Verapamil inhibited the transport of CQ via PfCRT (resistant phenotype) showing half-maximum inhibitory concentration of 30 ± 3 μM, as did a range of quinolines including quinine and amodiaquine. Hydrogen bond acceptor and two hydrophobic aromatic rings define the basic elements involved in interactions between PfCRT (resistant phenotype) and substrates or inhibitors [60].
Chemosensitizing acridinones were designed based on the critical features for CQ-chemosensitizing activity already mentioned (hydrogen bond acceptor and two hydrophobic aromatic rings). Firstly, Kelly et al. [35], synthetized and evaluated a series of 10-N-substituted acridinones bearing alkyl side chains with tertiary amino groups. The most potent CQ-chemosensitizer compound KF-A, 10-[6-(diethylamino)hexyl]acridin-9(10H)-one 28, Fig. 4, potentiated the antimalarial activity of CQ more than 80-fold in the Dd2 strain to a level (19 nM) similar to that observed for CQ-sensitive strain D6 (14,7 nM). CQ-chemosensitization effect was higher than those observed for verapamil and chlorfeniramine. Isobolar analysis of drug interactions revealed significant synergy. As was expected, acridinone bearing the N-10 alkyl chain but terminated with a chlorine atom, instead of terminal amino group, was without chemosensitization activity.
Later, the same authors, reported the discovery of the dual function acridinones which include, besides haem targeting structure, a chemosensitization moiety at the N10 position [8]. The most potent compound, T3.5 (27, Fig. 4), proved synergistic with chloroquine, amodiaquine, quinine or piperaquine but not with mefloquine against Dd2 strain. As was expected for verapamil-like chemosensitizers, no synergy between CQ and T3.5 (27, Fig. 4) was obtained for sensitive strain. Different behaviour, unlike verapamil, was observed for quinine synergy and PfCRT interaction, no explanation was established for those results.
The clinical utility of tricyclic antidepressants and antihistamine compounds as antimalarial chemosensitizers is limited by adverse effects at the high doses required for chemosensitizing efficacy. Since the antidepressants are believed to inhhibit neurotransmite uptake by the biogenic amine transporters and the antihistamine act by competing with histamine for H1 receptor sites on efector cells [35]. Although structural similarities between chemosensitizers acridinones and tricyclic antidepressants, no significant affinity with serotonin, dopamine or noradrenaline transporters was observed [35, 8].
CONCLUSIONS
Acridinone alkaloids represent the acridinone based structures with antimalarial activity isolated from natural sources. A few of those alkaloids have shown strong and selective antiplasmodial activity and have motivated synthesis of new structures. Several acridine and acridinone derivatives with antimalarial activity have been synthesised inspired by quinacrine (QA) and pyronaridine (Pyr) structures. Although recent studies have revealed potent acridine-based structures against susceptible and resistant strains, selectivity indexes remains moderated. The number of studies on synthetic antimalarial acridinones is low but this chemical family includes the most promising new compounds obtained so far. Inhibitory concentration fifty in the nanomolar range and selectivity indexes of thousands are the most relevant features. The most potent compounds exert their antimalarial action inhibiting hemozoin formation or mitochondrial bc1 Complex activity.

