All published articles of this journal are available on ScienceDirect.

Thiadiazine Derivatives as Antiprotozoal New Drugs
Authors Info & Affiliations
Abstract
The 3,5-disubstituted tetrahydro-2H-1,3,5-thiadiazine-2-thione scaffold have found many applications in recent years. This review is aimed at highlighting the most important aspects about these compounds: synthesis, spectroscopic characterization and antiprotozoan activities. How the chemical nature of N-substituents influences the overall activity / cytotoxicity profile will also be discussed.
1. INTRODUCTION
Trypanosomatids are parasitic protozoa responsible for several tropical diseases of which African sleeping sickness (Trypanosoma brucei gambiense and T. b. rhodesiense), Chagas disease (T. cruzi), and the different forms of leishmaniasis (Leishmania donoVani and L. infantum) are considered the most lethal [1]. At present, the chemotherapies associated with all forms of trypanosomiasis have many deficiencies, among them, poor efficacy, toxicity to humans, high cost, and the emergence of drug-resistant parasitic strains [2]. Unfortunately, because of the low profit margins associated with developing improved therapies for tropical diseases, there is no motivation for the pharmaceutical industry to develop better tropical disease drugs. Accordingly, many agencies, including the Special Program for Research and Training in Tropical Diseases at the World Health Organization (WHO/TDR), numerous academic research groups, various international/national bodies, and philanthropic foundations, have been sponsoring the discovery of new drug candidates for a number of tropical diseases [3].
Recently, the tetrahydro-2H-1,3,5-thiadiazine-2-thione (THTT) derivatives have received special attention due to the pharmacological properties that they display like antiparasitic agents. Although the 3,5-disubstituted tetrahydro-2H-1,3,5-thiadiazine-2-thione (mono-THTT) scaffold (Fig. 1) was synthesized for the first time in 1848 [4], its accurate structure was established in 1944 [5]. The first publications of these compounds pointed out the significance of the nature of the substituent at N-3 on antimicrobial activity [6], and toxicity of the substituent on N-5 has been shown [7]. Optimum activities had been obtained in compounds bearing lipophilic groups at the N-3 and hydrophilic ones at 5th position [8].
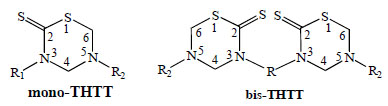
3,5-disubstituted tetrahydro-2H-1,3,5-thiadiazine-2-thione scaffold (mono-THTT) and alkyl-linked bis-(2-thioxo-[1,3,5] thiadiazinan-3-yl) carboxylic acids (bis-THTT).
The principal studies on the THTT derivatives [9-26] take into account the molecular structure of these molecules to obtain the best activity/cytotoxicity relation. In this context, we found in the bibliography the reports on many compounds with one THTT ring as the central core with a great variety of substituents in N-3 and N-5 position (mono-THTT) [9-18]. In the last years, to enhance the antiprotozoal effect two THTT rings were incorporated into the same molecular structure, connected to each other via their N-3 atom by a linear or branch aliphatic backbone and bearing alkyl or carboxyalkyl residues at N-5 (bis-THTT)[19-26] (Fig. 1).
Therefore this article will review the bibliography in order to show the development of new antiprotozoal agents using the THTT ring in their structure in the last ten years.
2. CHEMISTRY CONSIDERATIONS
2.1. 3,5 disubstituted-tetrahydro-2H-1,3,5-thiadiazine-2-thione (Mono-THTT)
The most used procedure to obtain the THTT derivatives is the reaction of the appropriate amine 1 with carbon disulfide 2 and potassium hydroxide 3, to give the dithiocarbamate potassium salt 4 (which was not isolated), followed by cyclocondensation with formaldehyde 5 and the selected amino acids [9-11, 13-15], pseudo peptides [9-11, 13-18], and amines or amino ester (6) [12] able to provide the nitrogen at 5th position of the thiadiazine ring in fair to moderate or good yields. In the first step of these synthetic procedures, water is employed as a protic polar solvent in order to stabilize the dithiocarbamate intermediate 4, in the second step pH 7-8 phosphate buffer was used (Scheme 1) [9-18].
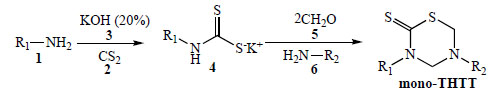
General procedure for obtaining mono THTT.
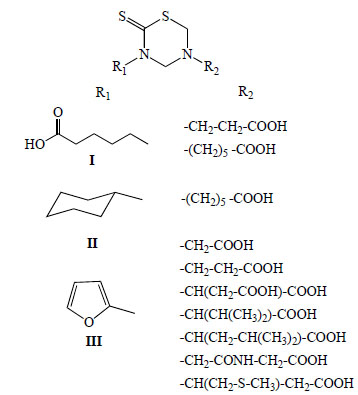
This methodology allowed the synthesis of 78 new mono-THTT derivatives to bunch up in at least seven series (Fig. 2). The series synthesized differ as to the nature (lipophilic or hydrophilic) of substituent at 3rd position. Some series bear an aromatic (furfuryl [9,11], benzyl [11] and D or L deacylated chloramphenicol [12]) and alkyl or cycloalkyl (ethyl, butyl, octyl, dodecyl, cyclopropyl and cyclohexyl) [9-11, 13-18] moiety, all of them lipophilic group; for other series the starting amines were hydrophilic (carboxyalkyl) group [9].
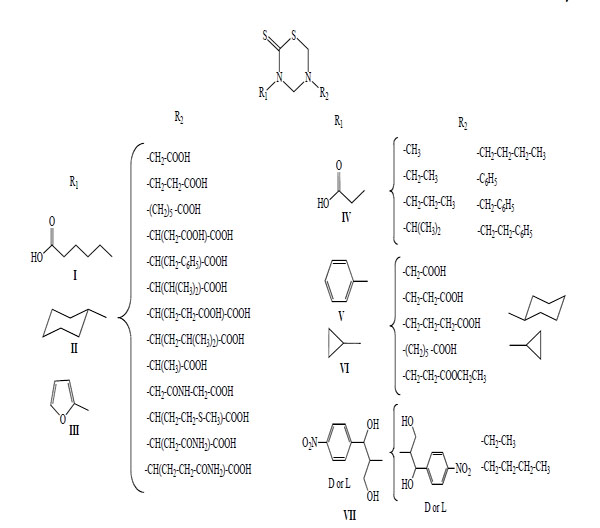
Representative mono-THTT synthesized.
Another synthetic method used to obtain these compounds was the solid phase synthesis of 3-(5-carboxypentyl)-5-substituted tetrahydro-2H-1,3,5-thiadiazin-2-thione derivatives (Serie I). 6-Amino-n-hexanoic acid was attached via its C-terminal to hydroxymethyl polystyrene using a ‘SASRIN’ linker. The bound amino acid was transformed into the corresponding dithiocarbamate 8 followed by cyclization in the presence of formaldehyde and the corresponding free amino acids to afford 3-(5´-carboxypentyl)-5-substituted tetrahydro-2H-1,3,5-thiadiazin-2-thiones (9). The final products I were cleaved from the resin and obtained in moderate yields due to low solubility of the corresponding free amino acids in 1,4 dioxan (Scheme 2).
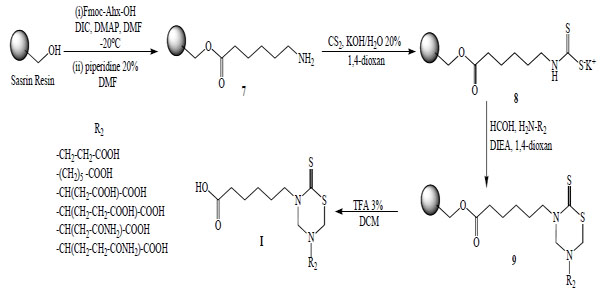
Solid Phase Synthesis of mono-THTT derivatives (I).
Although its use is limited by two factors: the possibility for the starting amines to be properly functionalized for the efficient coupling to the resin and the low solubility of the amino acids in the solvent employed. The development of this methodology would allow the generation of combinatorial library for THTT compounds.
2.2. Alkyl-linked bis-(2-thioxo-[1,3,5] thiadiazinan-3-yl) carboxylic acids (bis-THTT)
The synthetic methods for obtaining bis-THTT have been much less reported in the bibliography than the synthetic route for mono-THTT [19-22]. The general procedure is very similar to the one used for mono-THTT. The bis-THTTs were always obtained using diamines 10 and the amounts of all reagents were duplicated. In the first step the diamine reacts with carbon disulfide 2 in the presence of potassium hydroxide 3 to obtain the expected bisdithiocarbamate salt 11. The addition of formaldehyde 5 and the corresponding amine or aminoacid (6) to 11 resulted in the cyclocondensation in a slightly alkaline medium (phosphate buffer, pH 7-8) to generate, after treatment with HCl 15%, the desired bis-THTT (Scheme3) [19-21].
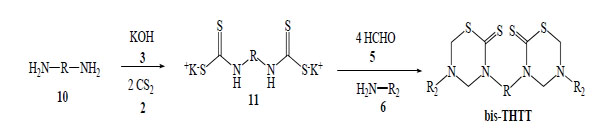
General procedure for obtaining bis-THTT synthesized.
This procedure allowed the synthesis of 43 new bis-THTT derivatives to bunch up in at least four series (Fig. 3). All compounds were obtained in moderate to good yields except those of series IX, probably due to the use of a bulky diamine (2,2-dimethyl-1,3-propanediamine) and the resulting steric hindrance at the cyclization stage.
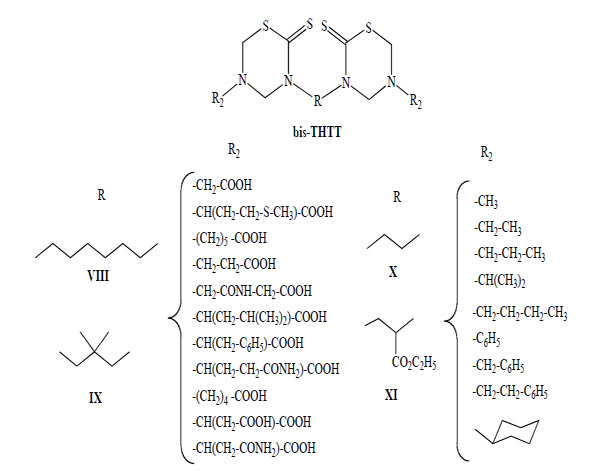
General structures of bis-THTT synthesized.
Recently, our group explored the feasibility of synthesis of new bis-THTT using more complex polyamines as linkage other than the initially reported diamines [22]. The N4-benzyl polyamine 14 was previously synthesized according to a methodology reported by O'Sullivan et al. [23] via a protection-deprotection strategy using ethyl trifluoroacetate as the selective protective group for primary amines in the presence of secondary amines. The synthetic route leading to derivatives of serie XII from the benzylated spermidine was similar to the one described above (Scheme 4).
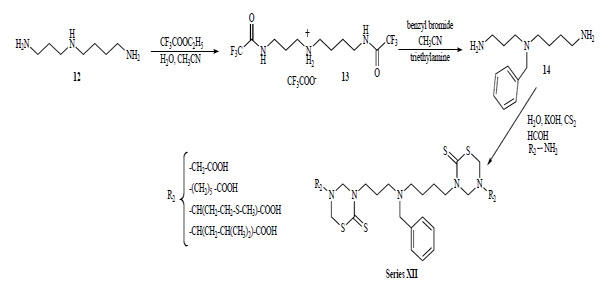
Synthetic route leading to bis-THTT (XII) from the benzylated spermidine.
The spermidyl linked bis-THTT series (XII) were obtained as solids in moderate yields.
The experimental procedure used to obtain the THTT derivatives (mono and bis-THTT) is simple, allowing a wealth of molecular diversity depending on the nature of groups attached to both nitrogens (N-3 and N-5) of the heterocycle.
2.3. Proposed Reaction Mechanism for the Generation of the Thiadiazine Ring
According to the authors, the formation of the thiadiazinane ring is performed via a one pot domino reaction between the pre-formed DTC, formaldehyde and the amino acid component. Despite being considered a multi-component reaction, the reactants are added in a stepwise fashion. Undoubtedly, one of the least explored aspects regarding the synthesis of THTT has been the study of the reaction pathway from the corresponding DTC. In one approach (Scheme 5a) the preformed DTC 4 is allowed to react simultaneously with formaldehyde and the corresponding amine to produce [substituted(aminomethyl)methanethionyl methylidenazanium 12 [20]. This species possesses two different reactive centers in the same molecular backbone, a protonated imine and a thiosulfanylmethylamino group. The intramolecular addition of the secondary amino group to the carbon atom of the methylidene moiety leads to the THTT ring. The formation of the THTT ring via a {[hydroxymethyl (substituted) carbamothioyl] sulfanyl}methanol intermediate 13 [13] was proposed (Scheme 5b). This process involves in situ generation of 13 from the corresponding DTC 4 and formaldehyde (5) followed by condensation with a primary amine. The isolation and characterization of an analogue of 13, via a crystallization process induced by the presence of KOH, was reported [13].
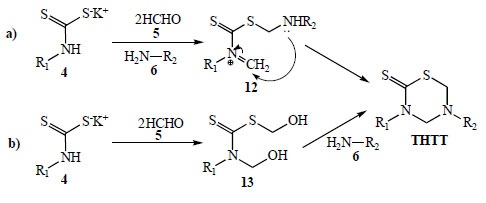
Proposed reaction mechanism for the generation of the THTT ring.
Recently, we have reported a preliminary DFT study aimed at predicting the probable cyclization mechanism of the thiadiazinane-2-thione from an intermediate of type 13 [24]. Based on experimental facts and DFT studies, a probable cyclization route to the THTT ring from the corresponding {[hydroxymethyl(substituted) carbamothioyl] sulfanyl}methanol intermediate 13 in aqueous medium is proposed. Notably, water not only contributes to the reaction as a mere solvent, but also plays an active role in the reaction mechanism. As far as we know, this is the first computational study on the formation of the thiadiazinane-2-thione ring.
3. STRUCTURAL CHARACTERIZATION OF MONO AND BIS-THTT DERIVATIVES
Structures of all the compounds were established on the basis of spectroscopic data and microanalysis. IR spectroscopy of the compounds showed absorption bands ranging from 3060 to 3450 cm−1 for (OH and NH), 1690 to 1720cm−1 for (C=O) and from 1445 to 1508 cm−1 for (C=S). Although, numerous studies have been published on the synthesis and characterization of these compounds [9-14], it was only in 2001 we published the first exhaustive structural characterization of mono-THTT derivatives [15-17]. These studies were considered an important structural data base to facilitate the characterization of novel compounds with the THTT ring in its structure.
The Nuclear Magnetic Resonance study deals with the complete 1H and 13C NMR assignments of a series of differently substituted THTT endowed with different organic addends on both heterocyclic nitrogen atoms (Fig. 4). The 1H NMR spectra at 300 MHz of THTT derivatives show, in general, two singlets corresponding to ring protons H-4 and H-6 around δ 4.50 and 4.40 respectively, besides other usual signals of the substituents. In order to assign unequivocally all NMR signals, 1D and 2D techniques such as DEPT (135), HMQC and HMBC were used [15].
Compounds studies by NMR 1D and 2D techniques.
A structural study of 5-carboxy-ethyl-3-(2´-furfurylmethyl) tetrahydro-2H-1,3,5-thiadiazine-2-thione was made by means of X-ray crystallographic analysis determining the most stable conformation in the solid state. The theoretical calculations allowed to gain a better picture of the conformational profile of the given compound by means of the semiempirical AM1 method, as well as by ab-initio calculations at Hartree-Fock level using 3.21G* and 6-31G* basis sets. 1H Noe experiments had also been carried out in order to obtain information about the conformational profile of this compound in solution [16].
The electrospray ionisation (ESI) in negative mode of the pharmacologically significant mono-THTT derivatives, and their subsequent fragmentations using an ion-trap mass spectrometer were investigated. Experiments on sequential product ion fragmentations (MSn) were performed in order to elucidate the degradation pathways for these compounds. The data reported show that the fragmentation of the even-electron M-H- ions could proceed through an internal nucleophilic substitution displacement. Decarboxylation and extrusion of carbon disulfide are other fragmentations observed [17].
On the other hand, the spectroscopic information gathered from previously synthesized mono-THTT derivatives [15-17] allowed to confirm the structure of the bis-THTT compounds. The structures of all the bis-THTT derivatives reported in the bibliography were established on the basis of spectroscopic data [19-22, 24, 25]. In general the 1H and 13C NMR signals of each THTT-ring were undistinguishable and all series, show a similar trend in the chemical shift of the common part of the molecular backbone. The 1H and 13C NMR spectroscopic data of alkyl, and polyamine-linked bis(2-thioxo-[1,3,5]thiadiazinan-3-yl) carboxylic acids, prepared from alkyl diamines and N4-(benzyl) spermidine, were fully assigned by the combination of one- and two-dimensional experiments (DEPT, HMBC, HMQC, COSY) [25]. In some cases, the 13C NMR experiments could not be performed because the bis-THTT derivatives were poorly soluble in DMSO at the concentration required for NMR spectroscopy [19, 20].
4. BIOLOGICAL ACTIVITY OF THTT DERIVATIVES
The promising results of antiprotozoal activity achieved by reported THTT derivatives could be attributed to the interaction of cysteine proteinases, present in most groups of parasitic protozoa [26], with isothiocyanates [27] generated by hydrolysis of the THTT ring in a protic medium [28]. Notwithstanding it should not be excluded the possible interaction of the released aminoacids or dipeptides, attached to position 5 of the THTT ring, with other molecular targets, enhancing the antiparasitic activity observed of these derivatives.
On the other hand, the high lipid solubility and ease of enzymatic hydrolysis [29] generally associated with this heterocycle, has motivated its use as a biolabile prodrug in the design of drug-delivery systems (DDS). The aforementioned properties and the possibility to attach several structurally distinct substituents to the heterocycle ring to modify either the biological or physico-chemical properties of these compounds, have prompted us to use the THTT ring as a template in many research programs aimed at the development of new antiparasitic compounds [9, 11-14, 18-22].
4.1. Biological Activity of Mono-THTT Derivatives
In 1999, we reported for the first time the antiprotozoan properties of 34 mono-THTT derivatives (Fig. 6) corresponding to the series I, II and III against Trichomonas vaginalis and Trypanosoma cruzi [9]. One year later, we published the study on the decomposition products of their mono-THTT and their anticancer properties (Fig. 5) [10].
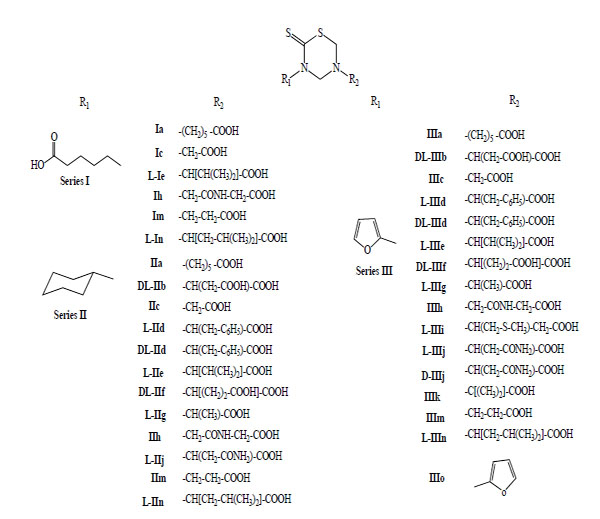
General structure of mono-THTT derivatives (Series I, II and III) assays like antiprotozoan drugs against Trichomonas vaginalis and Trypanosoma cruzi; and like anticancer drugs against HeLa, HT-29 and Hep G2 cells.
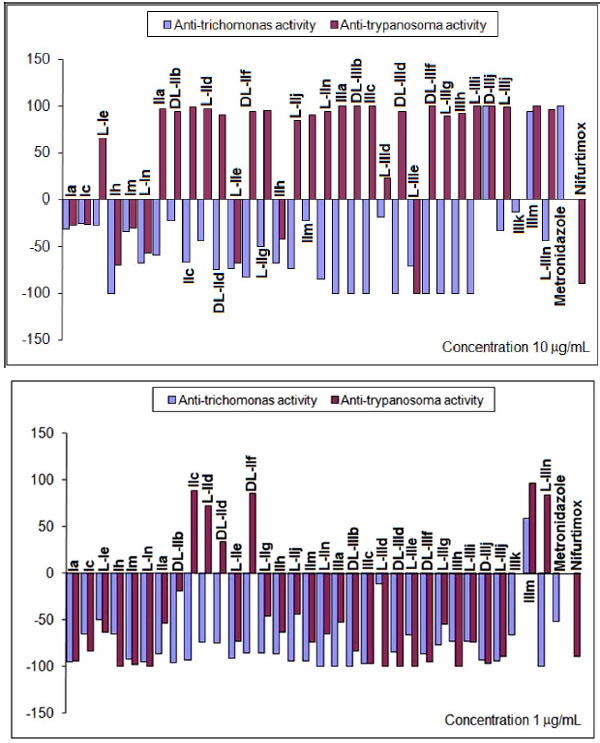
Percentages of reduction (positive) and/or growth (negative) at concentrations of 10 and 1μg/mL assayed in vitro against T. vaginalis and T. cruzi.
All mono-THTT derivatives studied showed significant in vitro antiprotozoan activity (both anti-trichomonas and anti-trypanosoma) at the highest dose tested (100 µg/mL), except for compound L-IIIe. (Fig. 6). The results obtained at 10 and 1µg/mL (Fig. 7) showed that the nature of substituents not afforded important differences in the trichomocidal activity (%R) of the three series studied, since most of the compounds lose their trichomonacidal activity at 10 µg/mL. This fact seems to indicate that the lipophilic character of R1 does not influence the in vitro trichomonacidal activity significantly. Only two compounds of series III (D-IIIj and IIIm) maintain their efficacy at 10 µg/mL and again IIIm was the most active compound at 1 µg/mL (%R=59), with a trichomonacydal effect similar to that of Metronidazole at 0.1 µg/mL (%R=52). None of the compounds studied were more active than Metronidazole (Fig. 6). The most active compounds, L-IIId, D-IIIj and IIIm, that were assayed in vivo, showed a reduction in the pathogenecity index, and this was mainly true for compound IIIm [9].
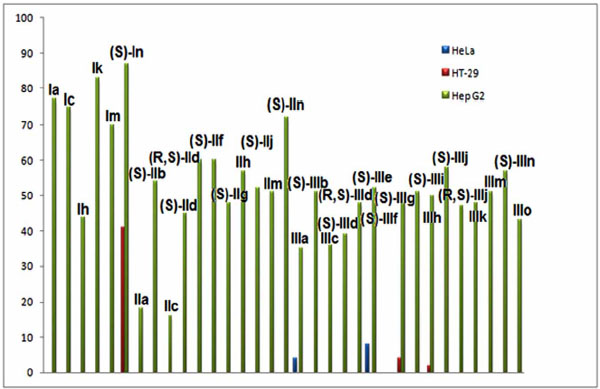
Cytotoxicity (IC50 (µmol/l)) of compounds belonging to series I, II and III against HeLa, HT-29 and Hep G2 cells.
Compounds of series II and III showed trypanosomicidal activity, both at 100 and 10 µg/mL, whilst compound of series I only showed cytostatic activity at 10 µg/mL. This fact indicated that the lipophilic substituents at the N-3 were better than hydrophilic ones for obtaining active compounds against T. cruzi. Compounds IIc, L-IId, DL-IId, DL-IIf, IIIm and L-IIIn maintained trypanosomicidal activity at 1 µg/mL showing a higher activity than Nifurtimox. We have found more compounds with trypanosomicidal than trichomonacidal activity (Fig. 6) [9].
Taking into account the highest citotoxic activities showed by some compounds of the series I, II and III in a previous work [9], we studied the anticancer properties of mono-THTT derivatives (Fig. 5). This study has been carried out by using cytotoxicity assays against HeLa, HT-29 and Hep G2 cells. The decomposition products of thiadiazinthione IIIm have been studied and their anticancer properties evaluated (Fig. 7) [10].
The results of this study could lead to the conclusion that most of the mono-THTT derivatives showed noticeable cytotoxic properties against HeLa and HT-29 but did not do so against Hep G2 cells. The compounds of series I and II were, in general, less cytotoxic than those of serie III, none of them showed a IC50 lower than 10 ìmol against any cell line. Compound IIIo, detected in the decomposition of compound IIIm and presumingly in one of the other thiadiazinones, were less cytotoxic than IIIm. The nature of R1 modulates the cytotoxicity of these compounds. Compounds bearing the aromatic furfuryl moiety (Series III) yielded the most interesting thiadiazinones. However, the nature of R2 influences the cytotoxic activity to a lesser grade. The (S) enantiomer showed more cytotoxic properties than its (R) isomer. Derivative (S) IIIj, bearing furfuryl and L-asparagine moieties, yielded the most interesting compound which is a candidate for a future anticancer study [10]. These results allowed the study through QSAR methodology of mono-THTT derivatives using the novel hybrid index pMRχ [30].
With the aim to increase the antiprotozoan activity spectra, our group reported new biological assays to study the non-specific toxicity and anti-amastigote activity of 18 of these compounds [31]. Cytotoxicity assays of 24 mono-THTT were performed. The 17 compounds with higher anti-epimastigote activity and lower cytotoxicity were, therafter, screened against amastigote of Trypanosoma cruzi. Out of these 17 derivatives, (S)-IId was selected to be assayed in vivo, because of its remarkable trypanocidal properties. Nifurtimox and benzidazole were used as reference drugs. All of the compounds were highly toxic at 100 µg/mL for macrophages and a few of them maintained this cytotoxicity even at 10 µg/mL. Of the derivatives assayed against amastigotes, Ik and (S)-IId showed an interesting activity, which was held even at 1 µg/mL. It is demonstrated that the high anti-epimastigote activity previously reported is mainly due to the non-specific toxicity of these compounds. In vivo assays assessed a reduction of parasitemia after the administration of (S)-IId to infected mice [31].
In 2004 and 2005 ten mono-THTT derivatives (Fig. 8) were tested in vitro for antiparasitic effects against both extracellular promastigotes and intracellular amastigotes of Leishmania amazonensis [32,33]. The compounds were found to be active against the amastigote form of the parasite, inhibiting parasite growing, from 10 to 89%, at a concentration of 100 µg/mL. This activity suggests that thiadiazine derivatives could be considered as potential antileishmanial compounds (Fig. 9) [32].
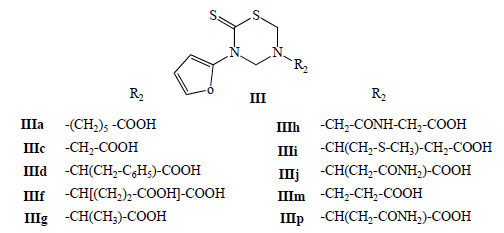
General structure of the mono-THTT (Series III) tested in vitro for antiparasitic effects against both extracellular promastigotes and intracellular amastigotes of Leishmania amazonensis.
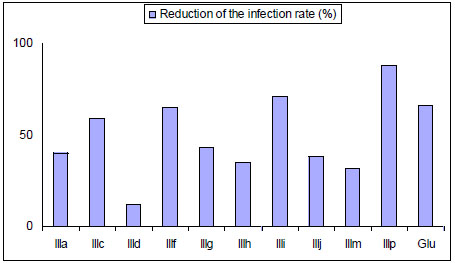
Effect of mono-THTT derivatives and Glucantine (Glu) on intracellular amastigote.
These results confirmed that the THTT represent a group of compounds with significant in vitro activity against L. amazonensis and suggested that some of them could be considered for further study as new therapeuthic alternatives [32,33].
All evaluated compounds caused an irreversible inhibition of the promastigote growth either after 1h of treatment with 10 µg/mL or after 24 h with 1 µg/mL (Fig. 10). However, the compounds exhibited high toxicity and produced inhibition of the phagocytosis in the murine host cell [33]. The mono–THTT tested exhibited a strong activity against L. amazonensis at low concentrations.
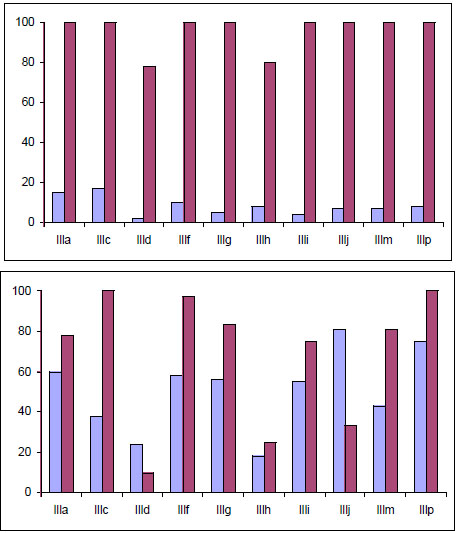
Effects of mono-THTT derivatives III on the L. amazonensis growth inhibition. A. Promastigotes were incubated in the presence of 10 µg/mL; after 1h of treatment (blue column); incubated 5 days in fresh medium (violet columns). B
4.2. Biological Activity of Bis-THTT Derivatives
It has been reported that bis-thiadiazines have antifungical, antimicrobial and antiprotozoal activity [19-22, 34]. Specifically with antiprotozoal activities three series of bis-THTT had been reported [21, 22, 34]. The in vitro activity of compounds belonging to series VIII and IXI (Fig. 11) against Leishmania donovani, Trypanosoma brucei rhodesiense, and Plasmodium falciparum was studied. From these results it may be observed (Fig. 12) that the best activity profiles were found against T. b. rhodesiense. It is intereresting that the activity against T. b. rhodesiense seems to be favoured for compounds having linear aminoacidic residues as substituents in N-5 of the THTT ring. Despite exerting a notable activity against T. b. rhodesiense, derivatives from series VIII appeared to be more citotoxic than the analogs of series IX (Fig. 13) [34].
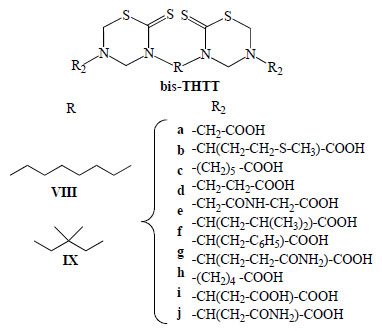
General structures of bis-THTT derivatives tested against Leishmania donovani, Trypanosoma brucei rhodesiense, and Plasmodium falciparum.
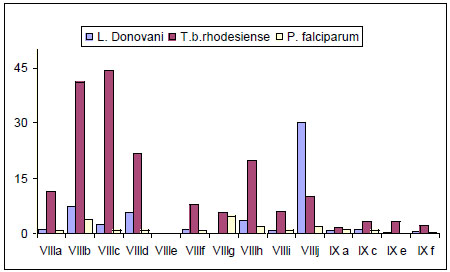
In vitro activity of compounds belonging to series VIII and IX against Leishmania donovani, Trypanosoma brucei rhodesiense, and Plasmodium falciparum.
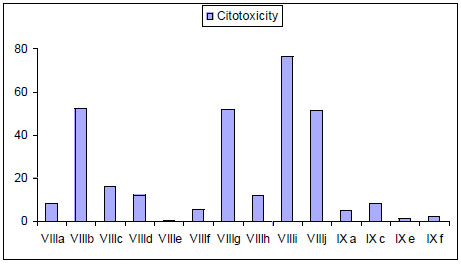
Cytotoxicity of bis-THTT of series VIII and IX.
The in-vitro activity against L. donovani, T. cruzi, T. b. rhodesiense , and P. falciparum of some compounds of series XII, was compared with these activities for compounds of series VIII (Fig. 14 and Fig. 15) [22]. For comparative purposes, structures VIII and XII were changed only in the nature of their connective backbone. In the bis-THTT derivatives in both series that had the same aminoacid residue (R2), the displayed protozoocidal profile against the three parasitic lines assayed was almost the same. However, higher values of therapeutic indexes were found for compounds VIIIa–c. Considering that both series exerted a similar protozoal activity it is possible to assume that structures XII are more cytotoxic than their alkyl tethers analogs [22].
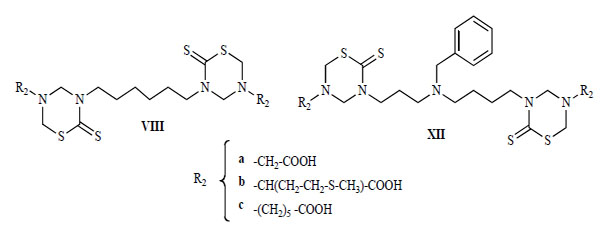
Bis-THTT structures (VIII and XII).
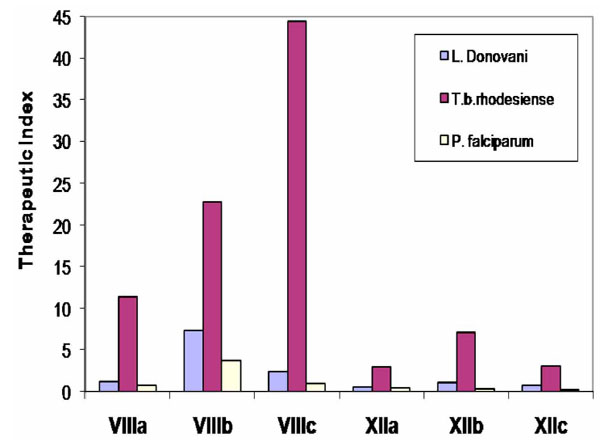
Results for in-vitro anti-protozoal activity of N4-(benzyl)spermidinyl-linked bis-THTT and their hexyl-linked bis-THTT analogs.
5. CONCLUSIONS
The most extensive method reported to obtain THTT derivatives proceed via a dithiocarbamate salt intermediary. This experimental procedure is simple and allows a wealth of molecular diversity depending on the nature of groups attached to both nitrogens of the heterocycle.
All THTT synthesized showed biological activity against some trypanosomatids at doses of 100 µg/mL, at low doses some of them showed important activities. The results presented in this review made it possible to analyze how the chemical nature of N-3 and/or N-5-substituents in the THTT ring influences the overall activity/cytotoxicity profile against some protozoa parasites. The introduction of two THTT rings in the same molecule potentiated the antiprotozoan activity of these derivatives.
The excellent properties of the THTT scaffold made its use as the main core in many projects for the development of new antiparasitic agents and the synthesis of novel bis-THTT derivatives employing other polyamines as bridging moieties will be also the theme of future investigations.

