All published articles of this journal are available on ScienceDirect.

Basic Aspects of the Pharmacodynamics of Tolperisone, A Widely Applicable Centrally Acting Muscle Relaxant
Abstract
Tolperisone (2-methyl-1-(4-methylphenyl)-3-piperidin-1-ylpropan-1-one hydro-chloride) was introduced in the clinical practice more than forty years ago and is still evaluated as a widely applicable compound in pathologically elevated skeletal muscle tone (spasticity) and related pains of different origin. In the present review, basic pharmacodynamic effects measured on whole animals, analyses of its actions on cell and tissue preparations and molecular mechanism of action on sodium and calcium channels are summarized as recently significantly new data were reported.
INTRODUCTION
The compound (Fig. 1) was synthesized as early as in 1956 as a result of research activity in Gedeon Richter Pharmaceutical Works. Tolperisone is an aryl alkyl β-aminoketone with an asymmetric carbon atom α to the carbonyl group.The dextrorotatory enantiomer was reported less effective, however no detailed analyses of the enantiomers are available [1]. The first pharmacological evaluation of its central anti-nicotine action without parasympatholythic activity was reported in 1958 by Nádor and Pórszász [2]. The first detailed characterization of its interneuron blocking activity on cats and rats was published by Pórszász et al. establishing that the compound is a specific inhibitor of the central nervous multineuronic synapses, capable to normalize the hyperreflexia elicited by intravenously administered 20 µg/kg strychnin which acts by blocking the collateral neurons of Renshaw, the inhibitory interneurons in the gray matter of the spinal cord [3].
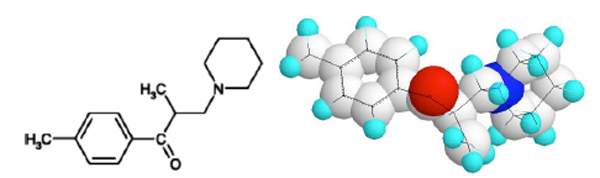
Chemical structure of tolperisone.
Tolperisonum hydrochloride (Mydeton, Mydocalm) was introduced in the clinical practice in the 1960’s with the primary indication to control muscular hypertonia. Nowadays it is in the market under several names (Musclex, Musolax, Mydosone, Tolson Plus, A-Calm, Besnoline F, Biocalm, Conspin, Lexaton, Miodom, Muscalm, Musocalm, Myderison, Myolax, Myoxan, Nichiperisone, Perilax, Quali-Docalm, Risocalm, Soneriper, Tanderon, Tolcalm, Tolesin, Tolfree, Tolperis, Tolpidol, Tolson, Topee, Toperin, Topeson, Topownan, Torimaron, Userm, Viveo). Within a few years following its introduction in clinical practice several reports were published on its acute and long-term efficacy in different rheumatological, orthopaedic, traumatological, neurological disorders (e.g. cerebral palsy, traumatic brain injury, stroke,multiple sclerosis and different forms of spinal cord injury), however only seven randomized clinical studies have been published.
CLINICAL TRIALS ON TOLPERISONE
The first randomized clinical trial on its efficacy and safety (Mydocalm, 300 mg, p.o. for 21 days) in the treatment of painful reflex muscle spasm in a prospective double-blind, placebo-controlled trial was published by Pratzel et al. [4] It was concluded that tolperisone hydrochloride represents an effective and safe treatment of painful reflex muscle spasm without the typical side effects of centrally active muscle relaxants.
In a randomized double blind and placebo controlled trial Mydocalm (150 mg twice daily for 12 weeks) was found to be a well tolerated and efficacious drug [5].
In a placebo-controlled double-blind clinical trial carried on a total of 72 healthy young adults single and repeated doses of Mydocalm (50 and 150 mg for 8 days) did not cause any sedation or impaired reaction times [6].
In a double-blind randomized crossover control study Bajaj et al. found that prophylactic intake of tolperisone hydrochloride (150 mg three times daily for 8 days) provides no relief to pain in course of post-exercise muscle soreness but results in reduction in isometric force [7].
The randomised, double-blind, placebo-controlled three-way cross-over study of tolperisone hydrochloride (300 mg, p.o.) and pridinol mesilate (8 mg, p.o.) on experimental jaw-muscle pain and jaw-stretch reflexes was studied by Svensson et al. It was concluded that, tolperisone provides a significant reduction in the perceived intensity of experimental jaw-muscle pain whereas had no effect on the short-latency jaw-stretch reflex [8].
Efficacy and safety of tolperisone (300-900 mg daily for 12 weeks) in the treatment of stroke-related spasticity in a randomized, double-blind, placebo-controlled, multicenter study with parallel groups was studied by Stamenova et al. [9]. Tolperisone reduced the mean Ashworth Score by a p < 0.0001 significance compared to the placebo group. Adverse events occurred less often on active treatment than on placebo and were mostly of mild-to-moderate intensity. Findings demonstrated the efficacy and excellent tolerance of tolperisone in the treatment of spastic hypertonia following cerebral stroke. It was also suggested that an individual dose titration which may exceed the recommended maximum dose of 450 mg daily results in optimized therapeutic benefit.
In a prospective, open-labelled, non-comparative, multi-centre observational, post marketing surveillance study conducted at 174 participating orthopaedic care centres across India 920 patients with painful muscle spasm associated with degenerative or inflammatory conditions were studied. Tolperisone (150 mg thrice daily for 7 days, p.o.) resulted in significant improvements (p < 0.0001) in Likert scale. No sedation was reported by any patient during study period and the incidence of common adverse effects like nausea, gastric irritation was less than 2% [10].
In a multicentric, randomized, comparative clinical trial, 250 eligible patients, with spasm of spinal muscles with acute or relapsing low back pain, of moderate to severe intensity and no finding of severe spinal diseases were randomly assigned to receive 150 mg of tolperisone thrice daily or 8 mg of thiocolchicoside twice daily for 7 days. The improvement in articular excursion on Lasegue’s maneuver was significantly greater on day 3 with tolperisone as compared to thiocolchicoside. The reduction in finger-to-floor distance score was greater on day 7 with tolperisone.The improvement in pain score at rest and on movement was significantly greater with tolperisone [11].
In the recently published consensus documented by Spanish Society of Neurology, Scottish Intercollegiate Guidelines Network system and German Neurological Society on recommendations for treatment of key symptoms such as spasticity in patients with multiple sclerosis tolperisone (as well as dantrolene and benzodiazepines) was evaluated as “not well documented” [12].
Tolperisone has been widely and effectively used in the clinical practice, however its exact mechanism of action has been not fully understood [13]. This might go back to the fact, that mechanisms involved in the pathophysiology of spasticity, one of several components of the upper motor neurone syndrome, have been and still are not sufficiently revealed [14].
PHARMACOLOGICAL DATA OF TOLPERISONE ON WHOLE ANIMALS
When monosynaptic and polysynaptic ventral root reflexes, the dorsal root potential and the dorsal root reflex were recorded in spinal cats, tolperisone (2.5-10 mg/kg i.v.) dose-dependently inhibited both ventral root reflexes and the dorsal root reflex, slightly prolonged the dorsal root potential without affecting the amplitude. However its effects were unique when compared its effects to baclofen and benzodiazepines (midazolam,diazepam) as these compounds abolished the monosynaptic reflex and only partially inhibited the polysynaptic reflex [15].
Participation of local anesthetic action (membrane stabilizing effect) in spinal reflex inhibition produced by tolperisone was examined by comparing the local anesthetic effects, the in situ depressant effects on muscle afferent discharges and the depressant effects on spinal reflexes in rats. The order of reducing afferent discharges by the drugs correspond on of their conduction blocking activities in the isolated sciatic nerve of rats. Membrane-stabilizing action in rats, spinal reflex inhibition and conduction blocking activity in the isolated sciatic nerve was reported by Ono et al. [16].
In anesthetized and laminectomized mice in the lumbo-sacral region, tolperisone transiently while baclofen long-lastingly inhibited the amplitudes of monosynaptic reflex potential giving further evidence on tolperisone’s membrane stabilizing activity with different mechanism of action of baclofen [17].
Tolperisone was reported to reversibly inhibit tonic periodontal massateric reflex in rats [18] and tonic masticatory neck reflex in patients [19].
In anesthetized intact rats tolperisone (5-10 mg/kg. i.v.) dose-dependently inhibited the group II flexor reflex reaching peak of about 50 and 75% at 2 min after administration and significant inhibition was observed within 30 min. The effects of chlorpromazine (0.2 and 0.6 mg/kg, i.v.) reached peaks of 50 and 80 %, respectively, at 5min after administration and significant inhibition was observed within 40 min. Baclofen (0.6, 1.3 and 2.5 mg/kg, i.v.) showed qualitatively and quantitatively similar effects. It was concluded that tolperisone, as a centrally acting muscle relaxant depresses group II flexor reflex by inhibiting descending noradrenergic tonic facilitation within the spinal cord [20].
Sakaue et al. [21] compared the effects of different drugs with sodium channel blocking effect used in the clinical practice for neuropathic pain on acute thermal and mechanical nociception. The following compounds were tested and assessed using the plantar and tail pressure tests: the antiepileptic carbamazepine and phenytoin, the centrally acting muscle relaxants tolperisone, zonisamide and eperisone, the local anaesthetic lidocaine and the µ-opioid receptor agonist morphine. Morphine produced analgesic effect both on acute thermal and mechanical nociception, while the other compounds including tolperisone (3-30 µmol/kg, s.c.) showed dose-dependent preferential elevation of the threshold for thermal nociception. This analgesic effect was correlated to their sodium channel blocking property. It was also shown in the sciatic nerve preparation isolated from mice that mexiletine, lidocaine, eperisone, and tolperisone impaired the propagation of low frequency action potentials, while carbamazepine, phenytoin and zonisamide action was obvious on higher frequency action potentials. Tolperisone was found to inhibit action potential propagation on both A- and C-fibers of rat sciatic nerve [22].
Muscle relaxant effect of toleprisone on spinal reflexes was first evidenced by Farkas et al. in1989, [15] however detailed analysis on the spinal reflex machinery between the stimulated dorsal root and the ventral root conveying efferent discharges in the rat spinal cord both in vitro and in vivo, in comparison with the local anesthetic lidocaine was performed by Kocsis et al. [23]. On isolated hemisected spinal cord preparation dorsal root stimulation-evoked ventral root potentials (DR-VRP) were recorded from the L5 ventral root. Tolperisone (50-400 µM) and its analogues (eperisone, lanperisone, inaperisone and silperisone) as well as lidocaine (200-800 µM) caused concentration-dependent depression of all studied components of DR-VRP. Significant attenuation of monosynaptic reflex potentials, afferent fiber potentials and excitatory postsynaptic potential were also produced by all the compounds studied.
In the in vivo spinal reflex study on decerebrated laminectomized rats all the compounds studied significantly attenuated monosynaptic, disynaptic, and polysynaptic reflex potentials. Tolperisone and eperisone had practically identical effects in the relative sizes of the effects and on the three different ventral root reflex components, however, peak effects of tolperisone were greater for all of the three components. The profile of lidocaine was different, producing weaker depressant effect on monosynaptic reflex potentials than on polysynaptic reflex potentials. Studying the afferent nerve conduction in vivo, possibility of a local anesthetic-type inhibition of afferent nerve conduction to the reflex inhibition following systemic silperisone, tolperisone, and lidocaine was compared. Tolperisone and silperisone left afferent nerve conduction practically unchanged, while lidocaine was slightly effective.
In decerebrated rats and mice tolperisone (10 mg/kg, i.v. or p.o.) and its analogues (eperisone and lanperisone) dose dependently depressed the muscle tone in the α-rigidity and γ-rigidity preparations, and the depression reached its maximum at 10 min following administration. Among the compounds studied only tolperisone decreased the frequency of muscle spindle discharges [24].
In the Spared Nerve Injury Model (SNIM) of neuropathic pain induced in rats effectiveness of tolperisone (10 mg/kg, i.p.) on thermal hyperalgesia and cold-allodynia evoked by various stimuli was similar to phenytoin (20 mg/kg) which is a standard compound as a sodium channel blocker. Results emphasise the concept that blockade of sodium channels by tolperisone may be beneficial for treatment of neuropathic pain [25].
ANALYSES OF TOLPERISONE’S ACTIONS ON CELL AND TISSUE PREPARATIONS
The first data on tolperisone’s effect on ionic currents was published by Hinck and Koppenhöfer [26]. Further analysis of tolperisone’s mechanism of effect using single intact Ranvier nodes of the toad Xenopus by means of the Hodgkin-Huxley formalism [27] showed that tolperisone (100 µM) decreased sodium permeability by about 50% in a potential-independent manner while other parameters of the sodium system remained unchanged. Potassium permeability was decreased only at strong depolarizing potentials and at week depolarization potential a significant increase in potassium permeability could be seen. All the effects were completely reversible. Apparent dissociation constants of 0.06 mmol/l and 0.32 mmol/l for sodium and potassium, respectively, were calculated. These findings clearly showed that tolperisone has fundamentally different effect on voltage gated sodium channel than local-anaesthetic compounds.
Quasthoff et al. [22] applied a new and elegant electrophysiological method for in vitro nerve compound action potential (CNAP) recordings of the isolated rat sural nerve including sodium channels functionally expressed in Xenopus oocytes. Two different Na+ channel subtypes α-subunits were functionally expressed, Nav1.6, that is mainly responsible for the A-fibre CNAP and the tetrodotoxin- resistant Nav1.8, mainly expressed in C-fibres [28]. Addition of tolperisone (100 mM) to the bath solution resulted in a fast and significant reductions both in myelinated A- and unmyelinated C-fibre compound action potential giving further evidence that tolperisone is an effective inhibitor of voltage-dependent sodium channels.
As tolperisone is structurally related to the sodium channel antagonists local anesthetics (lidocaine and procaine) [29] Hofer et al. [30] compared the acute effect of tolperisone and lidocaine on seven different isoforms of the voltage- dependent sodium channels (Nav1.2, Nav1.3, Nav1.4, Nav1.5, Nav1.6, Nav1.7, and Nav1.8 isoforms) expressed in the Xenopus laevis oocyte expression system. Sodium currents were recorded with the two electrode voltage clamp technique. Kinetic analysis of cumulative dose response curves showed that there were marked differences between the effects of the two drugs on identical isoforms, significant for the neuronal Nav1.6, Nav1.7 and Nav1.8 isoforms. The extent of use-independent (tonic) block (connected to pain treatment) on the same isoform was significantly stronger for tolperisone (Nav1.2, Nav1.3, Nav1.7 and Nav1.8). Both compounds affected recovery from inactivation. Tolperisone had no prolongating effect on fast recovery process, however lidocaine had prolongation of the time constant of the fast recovery process for the Nav1.3, Nav1.5, and Nav1.7 isoforms (characteristic for local anaesthetics).
Effects of tolperisone, silperisone, eperisone, and lidocaine on voltage-gated sodium currents on medium-sized dorsal root ganglion cells acutely dissociated from 6-day-old male rats was analysed by Kocsis et al. [23]. All the compounds studied inhibited peak sodium currents evoked by step depolarizations in a concentration-dependent manner, caused concentration-dependent shifts of the control inactivation curves toward the more hyperpolarized membrane potentials, but they also decreased the maximum available current. However at the IC50 concentrations for sodium channel blockade of the substances studied their N-type calcium channel blocker effect was very weak.
Comparing the effects of eperisone (analogue of tolperisone) and different Ca-antagonists on the basilar artery of the guinea pig [31] it was suggested that tolperisone may have Ca2+ channel antagonist effect. Novales-Li studied the effects of tolperisone, eperisone and isoperisone on the calcium current in an identified neurone of the snail Achatina fulica under voltage clamping [32], [33]. It was found that all the three drugs inhibited calcium current dose dependently without affecting activation time and shifted the steady state inactivation curves in the hyperpolarizing direction. Data suggested that the drugs do not bind to activated, but to resting and open Ca2+ channels.
We can conclude that the mechanism of action of the centrally acting muscle relaxant effect of tolperisone is related to its depressive effect on voltage-gated sodium and on a lesser extent on N-type calcium channels.
Tolperisone’s therapeutic effect is interpretable by its effective inhibiting action on afferent nociceptive input to the spinal cord, attenuation of spinal reflexes and inhibition of descending reticulo-spinal projections. However the effects of tolperisone in the brain were only recently analyzed [34], [35] using the robust, non-invasive technique of magnetic resonance imaging (MRI) for the direct observation of activation patterns evoked by tolperisone. The effect of tolperisone on the resting brain activity was studied in anaesthetized Wistar rats. To measure activation in the brain evoked by tolperisone the blood oxygen level dependent (BOLD) method of functional MRI (fMRI) was used. Effects of tolperisone were evaluated in prefrontal cortex, cingular cortex, primary somatosensory cortex, forelimb region, secondary somatosensory cortex, insular cortex,ventral posterolateral thalamic nucleus. Tolperisone (20 mg/kg i.p.) caused only non significant direct effect in the BOLD answer following its administration. However electrical stimulation of the rat forepaw caused massive BOLD increase in prefrontal cortex, cingular cortex , insular cortex, somatosensory cortex and thalamus and tolperisone almost completely prevented these provoked BOLD responses. Effect of tolperisone was most pronounced in the brain areas involved in pain sensation (prefrontal cortex, insula, thalamus and secondary somatosensory cortex). It is worth mentioning that lidocaine (10 mg/kg i.v.) was reported to rather increase painful train stimuli evoked BOLD responses in the brain [36], while an increased inhibitory effect of lidocaine (3 mg/kg i.v.) in the spinal cord on painful train stimuli evoked BOLD response increase was also reported [37].
There is a renewing interest in determination of the precise binding site of tolperisone on voltage-gated sodium channel. In a molecular modeling study [29] it was concluded that tolperisone and lidocaine can attach to identical protein binding sites, however no direct competition binding studies were still published. In the first pharmacological evaluation of tolperisone [2] tolperisone was shown to have central anti-nicotine action without parasympatholythic activity. On rat cortical synaptosomal preparation nicotinic acetylcholine receptor binding measured by [3H]BTX binding was inhibited dose dependently by tolperisone (IC50 40.9 ± 2.5 µM) and related centrally acting muscle relaxants (silperisone,eperisone, lanperisone,inaperisone) while lidocaine was one order of magnitude less potent [23].
SUMMARY
Tolperisone is a widely used compound in the clinical practice for pathologically elevated skeletal muscle tone (spasticity) and related pains. Tolperisone is a centrally acting muscle relaxant agent blocking spinal segmental reflex activity (Fig. 2). Recent evaluations of its mechanism of action revealed that the compound inhibits sodium channels and has N-type calcium channel blocking activity as well both in vitro and in vivo. Tolperisone is a specific inhibitor of the monosynaptic reflexes and only partially inhibits the polysynaptic reflexes. Its effects are unique compaired to baclofen and benzodiazepines, as it dose-dependently depresses group II flexor reflex by inhibiting descending noradrenergic tonic facilitation within the spinal cord. Tolperisone effectively inhibits afferent nociceptive input to the spinal cord, attenuates spinal reflexes and inhibits descending reticulo-spinal projections.Tolperisone inhibits action potential propagation on both in myelinated A- and in unmyelinated C-fibers and causes depression of all dorsal root stimulation-evoked ventral root potentials. Tolperisone has specific inhibitory effect on use-independent sodium channels (Nav1.2, Nav1.3, Nav1.7 and Nav1.8) connected to pain, which is a significant difference compared to lidocaine’s effect on sodium channels. Tolperisone’s effect was most pronounced in the brain areas involved in pain sensation (prefrontal cortex, insula, thalamus and secondary somatosensory cortex). Tolperisone hydrochloride represents an effective and safe treatment of painful reflex muscle spasm without the typical side effects of centrally active muscle relaxants.
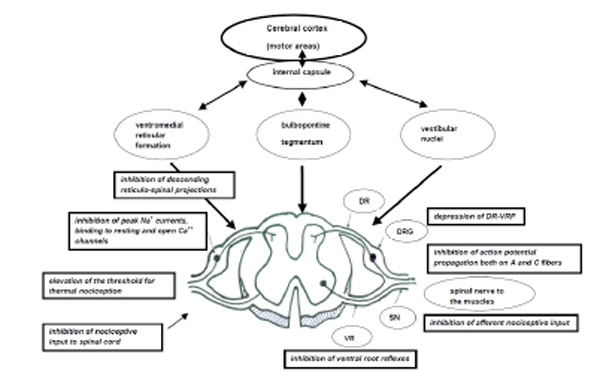
Mechanism of action of Tolperisone. (Abbreviations: DR: Dorsal root, DRG: Dorsal root ganglion, VR: Ventral root, SN: Spinal nerve, DR-VRP: Dorsal root stimulation-evoked ventral root potential).
CONFLICT OF INTEREST
Evaluation of literature data has solely been done by the author. Gedeon Richter Pharmaceutical Company sponsored the author and supplied some of the publications.
ACKNOWLEDGEMENTS
Advices and technical help of Andrea Melis and Zoltan Demeter are highly appreciated.
PATIENT’S CONSENT
Declared none.

