All published articles of this journal are available on ScienceDirect.

Synthesis and Biological Evaluation of Macrocyclized Betulin Derivatives as a Novel Class of Anti-HIV-1 Maturation Inhibitors
Authors Info & Affiliations
Abstract
A macrocycle provides diverse functionality and stereochemical complexity in a conformationally preorganized ring structure, and it occupies a unique chemical space in drug discovery. However, the synthetic challenge to access this structural class is high and hinders the exploration of macrocycles. In this study, efficient synthetic routes to macrocyclized betulin derivatives have been established. The macrocycle containing compounds showed equal potency compared to bevirimat in multiple HIV-1 antiviral assays. The synthesis and biological evaluation of this novel series of HIV-1 maturation inhibitors will be discussed.
1. INTRODUCTION
Currently, long-term suppression of viral replication with antiretroviral drugs is the only option for treating HIV-1 infection. To date, more than two-dozen antiretroviral drugs have been approved and shown to significantly increase patient’s survival. However, therapeutic regimens known as highly active antiretroviral therapy (HAART) are often complex because a combination of different drugs must be administered to patients to avoid the rapid emergence of drug-resistant HIV-1 variants. Furthermore, drug toxicity, adverse drug–drug interactions, and poor patient compliance can also lead to treatment failure. These will require continuous development of improved antiviral drugs with new targets and mechanisms of action [1].
The HIV Gag polyprotein precursor (Pr55Gag), which is composed of four protein domains [matrix (MA),capsid (CA = p24),nucleocapsid (NC), and p6] and two spacer peptides (SP1 and SP2), represents a new therapeutic target. Concomitant with particle release, the virally encoded protease (PR) cleaves Gag into four mature protein domains (MA, CA, NC and p6) and two spacer peptides (SP1 and SP2). Gag-Pol is also cleaved by PR, releasing the viral enzymes PR, reverse transcriptase (RT) and integrase (IN). Gag proteolytic processing, known as maturation, induces a morphological rearrangement within the particle [2]. Maturation converts the immature, donut-shaped particle to the mature virion, which contains a condensed conical core composed of a CA shell surrounding the viral RNA genome in a complex with NC and the viral enzymes RT and IN.
Bevirimat (PA-457, MPC-4326) is a maturation inhibitor that inhibits the final step in the processing of Gag (Fig. 1). It showed activity against ART-resistant and wild-type HIV-1, and demonstrated synergy with antiretroviral agents from all other classes. It was reported that bevirimat reduced HIV-1 viral load by a mean of 1.3-log copies/mL in patients who achieved through drug concentrations of 20 µg/mL and did not have any of the key baseline Gag polymorphisms at positions Q369, V370 or T371. The high baseline drug resistance due to the existing wild-type polymorphisms sets huge limitations to its clinical potential, and the further development of bevirimat was halted in June, 2010 [4].
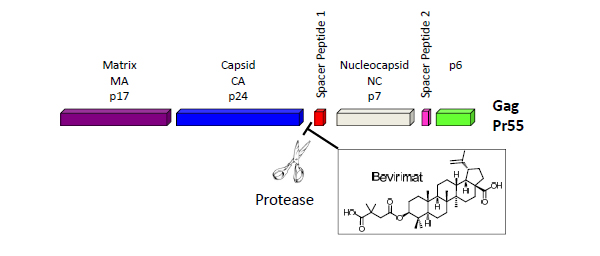
Cleavage at the CA–SP1 junction is the rate-limiting step in HIV-1 maturation, which is blocked by bevirimat [3].
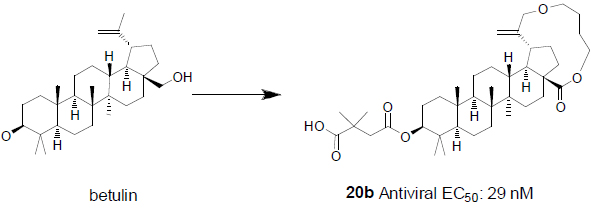
Inspired by the previous studies [5], we started an HIV-1 maturation program with the goal to obtain a better molecule with improved antiviral activity and minimal baseline polymorphism issue as mentioned above. Recently, plenty of efforts have focused on similar goals with various approaches [4, 6]. Due to insufficient knowledge on how to tackle the polymorphism issue, one approach we took was to build a macrocycle into betulin (Fig. 2). It has been well documented that macrocycles occupy a unique chemical space [7]. For any given scaffolds, macrocyclic compounds predominately originating from natural products often have profound pharmacological activity and selectivity by virtue of their diverse functionality and stereochemical complexity in a conformationally pre-organized ring structure. However, the synthetic challenge to access this structural class is high and hinders the exploration of macrocycles for drug discovery.
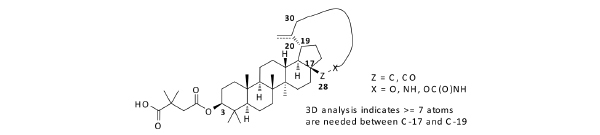
Design of macrocyclized betulin derivatives.
2. CHEMISTRY
In the past fifteen years, many bevirimat derivatives have been synthesized. However, the macrocyclization between C-17 and C-19 has not been explored. Synthetic routes to build the designed compounds have been tailor-made according to the size and functionalities within the macrocycles. As summarized in Scheme 1, commercially available betulin 1 was sequentially protected with a trityl group at the C-28 hydroxyl, and an acetyl group at the C-3 hydroxyl to give compound 2. The allylic oxidation of the isopropenyl group at the C-30 position of compound 2 with m-CPBA afforded allyl alcohol 3 in moderate yield. Alkylation of compound 3 with 1,5-dibromopentane gave the corresponding compound 4, from which the trityl group was deprotected with pyridinium p-toluenesulfonate (PPTS) and the liberated hydroxyl group was further converted to aldehyde by the treatment with 2-iodoxybenzoic acid (IBX). This aldehyde intermediate was then transformed to acid 5via a Pinnick Oxidation. Compound 5 was subsequently macrocyclized between the acid group at the C-28 position and the bromide to form compound 6. Following well established procedures, the final compound 20a was obtained by the deprotection of the acetyl group and the installation of 2,2-dimethyl-4-oxobutanoic acid at the C-3 position. The similar methodology was also used to make 20b and 20c.

Synthesis of compound 20a.
An alternative approach as depicted in Scheme 2 began with the alkylation of compound 3 with 2-bromoacetonitrile to give compound 7. Reduction of the nitrile with LAH afforded amine 8. Treatment of amine 8 with MsCl formed sulfonamide 9 in moderate yield, which was alkylated with 1,2-dibromoethane to extend its chain length. The acetyl group was introduced back to compound 10 at the C-3 position and Gabriel synthesis was then carried out to afford compound 11. The trityl group was removed with PPTS to give compound 12, in which the hydroxyl group at the C-28 position was transformed to acid. Hydrazine treatment exposed the amine in 13, and an amide coupling (HOBt/EDCI) afforded the desired macrocycle. The final compound 20d was obtained by subsequent deprotection of acetyl group and the installation of 2,2-dimethyl-4-oxobutanoic acid at the C-3 position. Similar methodology was employed to make 20e and 20f. Hydrogenation of compound 20d with PtO2 under 1 atm of H2 provided 20g; and 20h was obtained from 20f in the same way. The chirality at the C-20 position was not determined and the resulting product was considered as a diastereomer mixture.
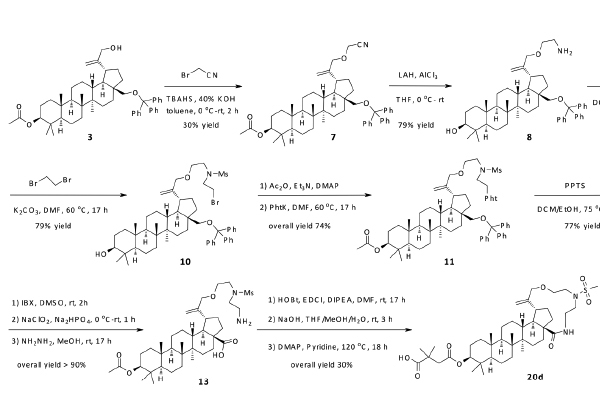
Synthesis of compounds 20d and 20g.
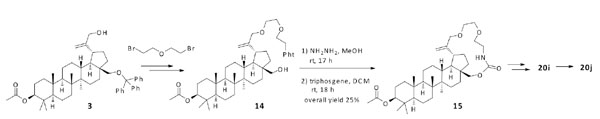
Synthesis of compounds 20i, and 20j.
The third macrocyclic system as shown in Scheme 3 was constructed in a related manner to that in Scheme 2. Compound 3 was reacted with 1-bromo-2-(2-bromoethoxy)ethane to extend its linker length, and Gabriel synthesis was carried out to give compound 14. After the treatment of compound 14 with hydrazine, macrocylization between the primary amine and hydroxyl group at the C-28 position with triphosgene yielded carbamide 15. The compound 20i was obtained by the deprotection of acetyl group and the installation of 2,2-dimethyl-4-oxobutanoic acid at the C-3 position. Further hydrogenation of compound 20i with H2/PtO2 furnished saturated analogue 20j as a diastereomer mixture.
Olefin metathesis was used as a final assembling strategy (Scheme 4). Allyl bromide was employed to make an extension at the C-30 position of compound 3. The resulting compound 16 was converted to compound 17 through similar approaches as described earlier. Subsequently, the carboxylic acid at the C-28 position was amidated with 6-hepten-1-amine to give compound 18, which was macrocyclized via olefin metathesis using Zhan 1B. During this process, the E/Z ratio of the newly formed double bond in compound 19 was not determined. Compound 20k was fully elaborated via similar steps as described earlier. The further reduction of the compound 20k using H2/Pd(OH)2 was unsuccessfully resulting in the ring opened product 20l in quantitative yield.
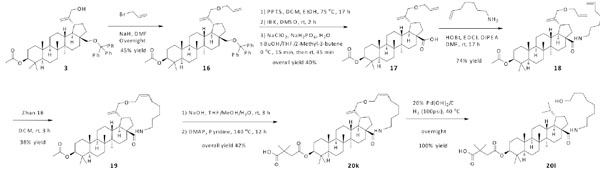
Synthesis of compounds 20k, 20l.
3. BIOLOGICAL ACTIVITY OF THE DERIVATIVES
All compounds and bevirimat were tested in HIV-1 lifecycle antiviral assay (Table 1) [8]. As shown in Table 1, macrocyclization modification of betulin is generally tolerated as multiple macrocyclized betulin derivatives (20b, 20d, 20g, 20h) demonstrated equal antiviral activities compared to bevirimat. The impact of the functionalities in the linker and the linker length remains unclear. For example, amide-linked compound 20f was more potent than the ester-linked compound 20c; whereas the amide-linked compound 20e was slightly less potent than the ester-linked compound 20b. Reduction of the double bond at the C-20 position showed either no impact on potency (20d and 20g) or slightly increase of potency (20f and 20h, 20i and 20j). The longer linker length decreased the potency (20a and 20b); however, the functionality within the linker also impacted this trend as shown by the comparison of compounds 20a, 20b, and 20d. The ring opening compound 20l actually showed high antiviral activity compared to its precursor 20k in this assay.
Anti-HIV-1 activity of compounds in HIV-1 lifecycle antiviral assay.
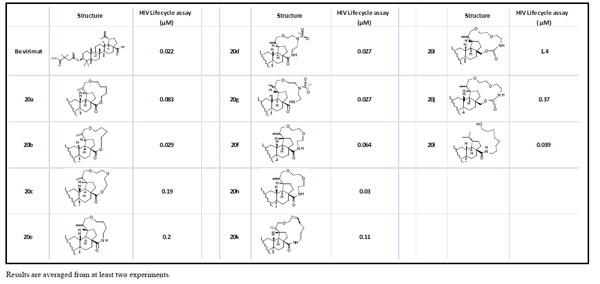 |
As one of the best macrocyclized betulin derivatives, compound 20b was selected to evaluate the effectiveness against a panel of clade B clinical isolates (Fig. 3, PBMC assay) [9]. ASJM108 carries the V370A polymorphism, which is highly resistant to bevirimat. Both bevirimat and 20b showed strong resistance to the V370A polymorphism strain ASJM108 in our hands. Macrocyclized 20b demonstrated better potency against a lab strain BA-L and virus BZ167 compared to bevirimat; however, it exhibited less effectiveness against virus BK132. Overall, the results revealed that bevirimat and compound 20b had a similar isolates coverage profile in this panel. Optimal structural requirement(s) for inhibition of a huge panel of clinical isolates (clade A, B, C etc.) remains a challenge and further optimization is required.
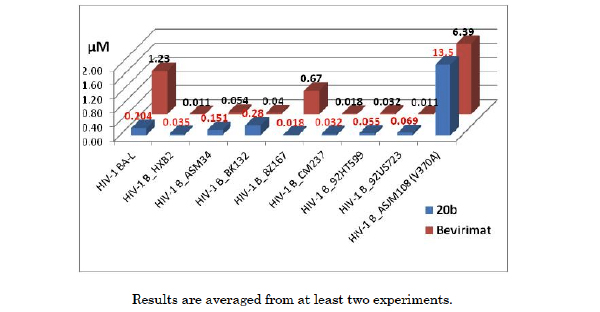
Activity of bevirimat and 20b against a panel of clade B clinical isolates.
CONCLUSION
In summary, efficient synthetic routes to macrocyclized betulin derivatives were established and these derivatives were evaluated biologically as anti-HIV-1 agents. Our macrocyclization approach successfully generated a novel class of HIV-1 maturation inhibitors. Although these macrocyclized betulin derivatives demonstrated similar potencies as bevirimat against wide-type V370 viruses, this new class of analogues did not efficiently overcome SP1 polymorphism issue when a limited panel of clade B isolates were explored. While we were unsuccesfull in the discovery of a clinically developable molecule by this route, we have investigaged other methods towards the aim of which will be reported.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
PATIENT’S CONSENT
Declared none.
ACKNOWLEDGEMENTS
Advices and technical help of Andrea Melis and Zoltan Demeter are highly appreciated.
REFERENCES
(b) Lataillade, M; Chiarella, J; Yang, J; Schnittman, R; Wirtz, S; Uy, V; Seekins, J; Krystal, D; Mancini, M; McGrath, M; Simen, D; Egholm, B Prevalence and clinical significance of HIV drug resistance mutations by ultra-deep sequencing in antiretroviral-naïve subjects in the CASTLE study PLoS One, 2010, 5(6), e10952.(c) Jakobsen, M E; Jakobsen, M R; Tolstrup, M; Søgaard, O S; Jørgensen, L B; Gorry, P R; Laursen, A; Ostergaard, L Transmission of HIV-1 drug-resistant variants: Prevalence and effect on treatment outcome Clin Infect Dis, 2010, 50, 566-573.
(b) Zhou, J; Yuan, X; Dismuke, D; Forshey, B M; Lundquist, C; Lee, K H; Aiken, C; Chen, C H Small-molecule inhibition of human immunodeficiency virus type 1 replication by specific targeting of the final step of virion maturation J Virol, 2004, 78(2), 922-929.
(b) Coric, P; Turcaud, S; Souquet, F; Briant, L; Gay, B; Royer, J; Chazal, N; Bouaziz, S Synthesis and biological evaluation of a new derivative of bevirimat that targets the Gag CA-SP1 cleavage site Eur J Med Chem, 2013, 62, 453-465.
(b) Marsault, E; Peterson, L M Macrocycles are great cycles: applications, opportunities, and challenges of synthetic macrocycles in drug discovery J Med Chem, 2011, 54(7), 1961-2004.

