All published articles of this journal are available on ScienceDirect.

One-pot Synthesis of Chemical Delivery Systems for Gamma-aminobutyric Acid and its Analogues
Authors Info & Affiliations
Abstract
Introduction:
A one-pot synthesis route was proposed for the synthesis of chemical delivery systems of bifunctional compounds containing carboxylic- and amino groups, such as gamma-aminobutyric acid and its structural analogues, gabapentin and phenibut.
Methods:
To obtain targeted compounds, consecutive esterification, amidation, methylation, and reduction were employed in contrast to a more traditional approach that requires isolation and purification of semi-products after each reaction step.
Results:
A significant (nearly two-fold) increase in yields was observed when target compounds were synthesized using a one-pot approach compared to the conventional one.
Conclusion:
One-pot approach is a possible and beneficial way for the synthesis of chemical delivery systems for gabapentinoids and other drugs containing carboxylic and amino groups.
1. INTRODUCTION
Drug delivery to the central nervous system is a major problem that has long drawn the attention of researchers in various fields [1-3]. The main reason for this is the presence of a specialized cellular structure in blood capillaries, the blood-brain barrier (BBB), which is effectively a semipermeable membrane that excludes most solutes that are present or might be present in the blood [4]. Many polar compounds and those of high molecular weight are unable to diffuse the cellular membrane and many lipophilic molecules that are able to do so are pumped back into the bloodstream as a result of interaction with specialized efflux receptors, such as multidrug resistance proteins [5]. There are several ways to bypass the BBB in order to treat a certain condition. First, with some conditions, such as strokes and cancer, BBB may be (transiently) opened [6]. However, those openings often occur in suboptimal temporal windows and cannot be relied upon for therapeutic needs, for e.g., in the case of ischemic stroke [7], or those do not result in homogeneous drug delivery to the desired areas of the brain [8]. Second, BBB can be disrupted by deliberate physical disturbances, such as ultrasound [9] or hyperosmotic solution [10]. However, this is a risky strategy that involves many side effects from other species found in the blood that enter the brain parenchyma in the absence of a protective barrier [11]. Third, a barrier can be traversed by means of colloidal drug carriers of heterogeneous nature [12]. Finally, there is a chemical approach to drug delivery to the brain. It implies reversibly changing the chemical structure of a drug so that it gains certain qualities that allow it to pass through the BBB, for instance, by making it able to bind to endocytosis receptors (active transport) or by imparting higher lipophilicity while at the same time ensuring that the new molecule will not be a substrate to multiple drug resistance receptors (passive transport) [13]; such modified drugs are known as “prodrugs.” Similar to the prodrug strategy is the concept of chemical delivery systems (CDS), which utilizes known metabolic processes (retrometabolic approach) for site-specific delivery of drugs, and relies on using enzymatic redox reactions to increase their accumulation in the brain parenchyma [14]. Usually, it is achieved by using substrates of NADH dehydrogenase, such as dihydropyridines, which, while reduced, are non-charged and lipophilic, and when oxidized, are charged and more hydrophilic, and thus unable to cross the BBB back into the bloodstream (are “locked-in”) [15].
Usually, such transformations require several steps, especially when dealing with bifunctional molecules, such as amino acids. Therefore, devising a synthetic route that allows obtaining the target compounds with increased yield appears to have practical value. In the past few decades, a one-pot approach for multistage syntheses has been gaining popularity due to its promise of higher yields and relative simplicity [16]. The term “one-pot” refers to a variety of approaches, such as cascade, telescope, domino reactions, etc. For the purposes of this research, by “one-pot”, we mean a telescopic approach to synthesis, one that allows evaporation, extraction, and filtration (with the target compound remaining in solution) without the isolation of intermediate products [17]. This strategy was applied to produce chemical drug delivery systems of gamma-aminobutyric acid and its analogues, and it has resulted in relatively high yields.
2. MATERIALS AND METHODS
Gamma-aminobutyric acid hydrochloride was provided by Acros Organics, gabapentin hydrochloride was purchased from Henan Famo Technology Co., Ltd., and phenibut hydroch- loride was a gift from Mir-Pharm LLC. All other reagents were of the highest quality available, and were used without additional purification. Silica gel (0.04–0.063 mm/230–400 mesh) for column chromatography was purchased from Macherey-Nagel GmbH & Co KG.
2.1. General Procedure
To an ice-cold suspension of a gabapentinoid (GABA, gabapentin, phenibut, I) in the form of hydrochloride in methylene chloride, a catalytic amount (5 mol%) of DMF was added, followed by 2 eq. of oxalyl chloride in an inert atmosphere of argon. Over the period of 1 h, temperature was slowly raised, and then the mixture was refluxed for two additional hours (until the bubbling subsided). Methylene chloride and excess oxalyl chloride were removed in vacuo, and in the case of low-boiling alcohols, a large excess (25 ml) of the alcohol was added. In the case of high-boiling alcohols, (1.1 equivalent) was dissolved in a fresh portion of methylene chloride, and the solution was then added to a reaction mixture. The reaction continued overnight, yielding an ester II.
To a solution of II in methylene chloride in an ice bath, 1.2 eq. of nicotinoyl chloride hydrochloride was added, followed by 3 eq. of triethylamine. The reaction was allowed to proceed overnight, and then methylene chloride was removed in vacuo and replaced with tetrahydrofuran in order to remove the triethylamine hydrochloride by filtration.
To the filtrate containing III, 4 eq. of methyl iodide was added, and the reaction mixture was refluxed for 12 hours. After the reaction was completed, tetrahydrofuran was removed; the residue was dissolved in methylene chloride, cooled down on an ice bath, and then deaerated water, sodium bicarbonate (4 eq.), and sodium dithionite (3 eq.) were added. The reaction proceeded for 15 min in an ice bath, and then the bath was removed and the reaction was proceeded for additional 30 minutes. Then, the layers were separated, and the aqueous layer was extracted with methylene chloride, combined and washed with deaerated water, dried over sodium sulfate, and evaporated in vacuo. The resultant viscous reddish oil was stored at −20°C in the atmosphere of argon, protected from light.
In order to remove minute impurities, vacuum liquid chromatography was applied; the eluent used was isopropyl alcohol-methylene chloride (gradient 0 ‒ 5 vol.%), with the constant addition of 0.5% triethylamine in order to deactivate silanol groups and prevent the substance from being stuck on the silica.
3. RESULTS AND DISCUSSION
Retrametabolic drug design in general, and chemical delivery systems in particular, have been applied to various drugs in order to allow them to bypass the BBB [18]. One of the key differences between retrametabolic drug design and more conventional prodrug design is the number of stages a molecule undertakes before being reversed to the original active pharmaceutical compound possessing the necessary therapeutic effect. Therefore, it often means that the original molecule must undergo several steps of chemical modification. Naturally, the most efficient route is desired for the process to be economically and environmentally sound.
In the present study, we have selected several gamma amino acids, such as gamma-aminobutyric acid and its structural analogues, gabapentin and phenibut (Fig. 1), as candidates for modification. Chemical delivery systems have already been obtained for GABA and gabapentin [19, 20], but so far, no such derivatives have been created for phenibut. We foresee that the approach presented here can also be used for other biologically active molecules possessing carboxylic and amine functionalities, such as alpha- and beta-amino acids, short peptides, and other drugs that are not classified as amino acids.
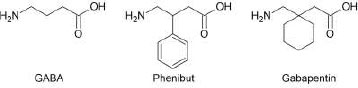
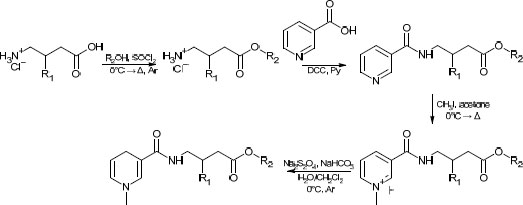
In several studies [19, 20] aimed at the creation of the CDS for gabapentinoids, yields either have not been reported, or in the case of CDS for GABA containing a benzyl moiety [20], have been similar to the one we have obtained in the present study. For the purposes of comparison of the synthetic routes, the method described in the aforementioned papers was recreated in our lab. The former route is presented in Scheme 1.
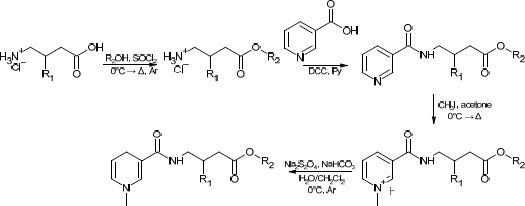
While facile, this route has several drawbacks, such as low atom economy, use of rather toxic pyridine, and overall low yields (typically in the range of 25 to 35%). The route proposed in this study can be found in Scheme 2. One major concern was its applicability to various alcoholic moieties; therefore, relatively low-boiling alcohols (methanol, n-butanol) and high-boiling alcohols (cyclohexanol, benzylic alcohol) were applied. It was found that the addition of a near-stoichiometric amount of high-boiling alcohol and allowing it to react with acyl chloride under reduced pressure did not significantly lower the overall yield compared to low-boiling alcohols. The slight excess was taken into account in the second stage (amidation with nicotinoyl chloride) by a corresponding increase in the amount of the reagent. This bore little significance at the latter stages, which required a significant excess of reagents.
The yields of the synthesized compounds are given in Table 1.
| No. | Original Compound | Alcohol Moiety (R2) |
Total Yield, % | |
|---|---|---|---|---|
| Conventional Synthesis | One-pot Synthesis | |||
| 1 | GABA | isopropyl | 23 | 44 |
| 2 | GABA | benzyl | 33 | 59 |
| 3 | Gabapentin | isopropyl | 24 | 76 |
| 4 | Gabapentin | n-butyl | 28 | 67 |
| 5 | Gabapentin | cyclohexyl | 33 | 63 |
| 6 | Gabapentin | benzyl | 25 | 70 |
| 7 | Phenibut | isopropyl | 38 | 58 |
| 8 | Phenibut | benzyl | 27 | 55 |
CONCLUSION
Despite the necessity to purify the final product using chromatography, yields were still higher than those using the conventional synthetic approach. The proposed one-pot synthetic route has allowed us to obtain the target compounds in yields about twice as high compared to the conventional approach, and has also achieved a better atom economy, thus bringing it closer to the compliance with principles of green chemistry. We suggest that the route presented in this study may be used for other compounds bearing dual functionalities, viz., carboxylic and amine groups, such as amino acids and short peptides.
LIST OF ABBREVIATIONS
| BBB | = Blood-brain Barrier |
| CDS | = Chemical Delivery Systems |
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
No humans or animals were used for the studies that are the basis of this research.
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIAL
All the data supporting the findings can be found in the paper itself, there is no need for an external repository.
FUNDING
This work was supported by a grant from the Russian Fund for Basic Research (RFBR) with No. 20-015-00220.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGEMENTS
Declared none.
SUPPLEMENTARY MATERIAL
1H NMR spectra were recorded using a Bruker AC 400 spectrometer with a working frequency of 400.13 MHz; the internal standard was TMS. All spectra were recorded in DMSO-d6.
Isopropyl 4-(1-methyl-1,4-dihydropyridine-3-carboxamido) butanoate (1)
1H NMR: δ 1.31 (6H, d, J = 6.9 Hz, CH3), 1.87 (2H, m, CH2), 2.65 (4H, m, CH2CO, CH2NH), 2.80 (1H, s, CH3), 3.20 (2H, t, J = 7.5 Hz, CH2), 4.59 (1H, m, CHpyr), 5.02 (1H, m, CH), 6.81 (1H, m, CHpyr), 7.93 (1H, s, CHpyr).
Benzyl 4-(1-methyl-1,4-dihydropyridine-3-carboxamido) butanoate (2)
1H NMR: δ 1.77 (2H, m, CH2), 2.53 (2H, t, J = 7.4 Hz, CH2pyr), 2.68 (2H, m, CH3), 2.77 (2H, t, J = 7.5 Hz, CH2CO), 3.36 (2H, t, J = 7.8 Hz, CH2NH), 4.60 (1H, m, CHpyr), 5.14 (2H, s, CH2), 5.78 (1H, m, CHpyr), 6.86 (1H, m, CHpyr), 7.28 (s, CHar).
Isopropyl 2-(1-((1-methyl-1,4-dihydropyridine-3-carboxamido)methyl)cyclohexyl)acetate (3)
1H NMR: δ 1.02-1.60 (16H m, CH2chex, CH3), 2.36 (2H, CH2CO, s), 2.62 (2H, m, CH2Pyr), 2.84 (3H, s, CH3), 3.36 (3H, s, CH2NH), 4.79 (1H, m, CHPyr), 5.02 (1H, m, CH), 6.78 (1H, m, CHPyr).
n-Butyl 2-(1-((1-methyl-1,4-dihydropyridine-3-carboxamido)methyl)cyclohexyl)acetate (4)
1H NMR: δ 0.88 (3H, t, J = 7.1 Hz, CH3), 1.04-1.62 (12H, m, CH2, CH2chex), 2.35 (1H, s, CH2CO), 2.64 (2H, m, CH2pyr), 2.78 (2H, s, CH3), 3.23 (2H, s, CH2NH), 4.10 (2H, t, J = 7.1 Hz, CH2O), 4.70 (1H, (1H, m, CHPyr),5.87 (1H, m, CHPyr),6.90 (1H, m, CHPyr).
Cyclohexyl 2-(1-((1-methyl-1,4-dihydropyridine-3-carboxamido)methyl)cyclohexyl)acetate (5)
1H NMR: δ 1.08-1.80 (20H, m, CH2chex), 2.30 (2H, s, CH2CO), 2.64 (2H, m, CH2pyr), 2.82 (3H, s, CH3), 3.23 (2H, s, CH2NH), 4.65 (1H, m, CHPyr), 5.79 (1H, m, CHPyr),6.84 (1H, m, CHPyr).
Benzyl 2-(1-((1-methyl-1,4-dihydropyridine-3-carboxamido)methyl)cyclohexyl)acetate (6)
1H NMR: δ 0.95-1.60 (0H, m, CH2chex), 2.36 (2H, s, CH2CO), 2.53 (2H, m, CH2pyr), 2.78 (3H, s, CH3), 3.36 (2H, s, CH2NH), 4.52 (1H, m, CHpyr), 5.14 (2H, s, CH2), 5.86 (1H, m, CHpyr), 6.66 (1H, m, CHpyr), 7.46 (5H, s, CHar).
Isopropyl 4-(1-methyl-1,4-dihydropyridine-3-carboxamido)-3-phenylbutanoate (7)
1H NMR: δ 1.31 (6H, d, J = 6.9 Hz, CH3), 2.45 (2H, d, J = 6.9 Hz, COCH2), 2.60 (2H, m, CH2Pyr), 2.82 (1H, s, CH3), 3.30 (2H, m, CH2NH), 4.53 (1H, m, CHPyr), 4.87 (1H, m, CHPyr), 5.85 (1H, m, CHPyr), 7.05-7.20 (5H, m, CHAr)
Benzyl 4-(1-methyl-1,4-dihydropyridine-3-carboxamido)-3-phenylbutanoate (8)
1H NMR: δ 2.33 (2H, m, COCH2), 2.67 (2H, m, CH2Pyr), 2.80 (1H, s, CH3), 3.40 (2H, m, CH2NH), 3.30 (2H, m, CH2NH), 4.47 (1H, m, CHPyr), 5.13 (1H, s, CH2O) 5.55 (1H, m, CHPyr), 6.75 (1H, m, CHPyr), 7.15-7.21 (5H, m, CHAr).

